Figura 1
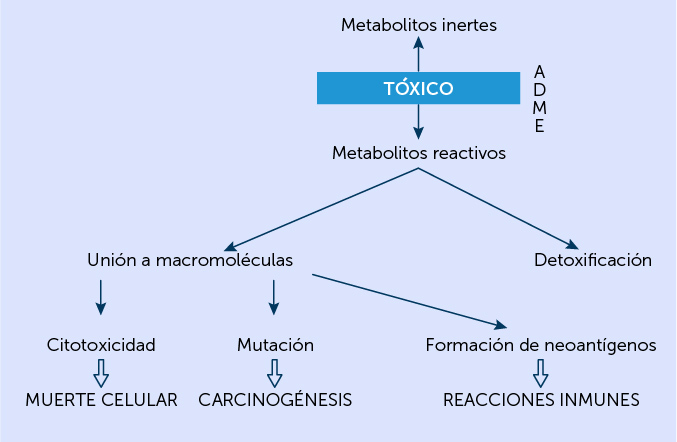
Resumen de la Toxicocinética y la Toxicodinamia de los compuestos químicos en el organismo. (ADME, Absorción, Distribución, Metabolismo, Eliminación.)
Coordinadora: Dra. Rubí Hernández Sánchez
LA CIENCIA DE LA TOXICOLOGÍA
Dr. Miguel Ángel Montoya Cabrera, Dra. Rubí Hernández Sánchez
El origen de la farmacología y la toxicología tiene un tronco común: la herbolaria misma, de la que hay evidencias que era practicada por las culturas primitivas. El conocimiento de que algunas plantas causaban efectos deletéreos fue aprovechado por el hombre primitivo quien impregnaba las puntas de sus flechas con los extractos de dichas plantas, para de esta manera aumentar el daño a sus presas de caza o sus semejantes. De ahí el origen del vocablo tóxico, que proviene del griego que significa precisamente "flecha".
Más tarde se extendió el conocimiento de los tóxicos a aquellos de otro origen que no fueran los vegetales o sus frutos. Así, en el papiro de Ebers (1500 a.C.), ya se hace referencia a venenos animales, a metales y se identifican algunas plantas venenosas que no han perdido actualidad como la cicuta, el acónito y el opio. Dioscórides elabora la primera clasificación conocida de los tóxicos según su origen: plantas, animales y minerales, que con algunas variaciones es casi la misma clasificación empleada en la actualidad. Con un pensamiento más mágico que científico, a varios de los tóxicos se les atribuyeron propiedades supuestamente afrodisiacas.
Con estos fines se elaboraban pociones frecuentemente conocidas como "pociones de amor de Venus", siendo éste el origen de otro vocablo relacionado con estas sustancias, el “veneno". En el momento actual se conservan ambos términos, pero obviamente con connotaciones diferentes. Así, un tóxico se define como una sustancia de naturaleza química y origen fuera del organismo (de ahí que también se les designe como xenobióticos), que introducido en el mismo y dependiendo de su dosis y tiempo de acción, va a actuar sobre sistemas biológicos específicos, dando lugar a alteraciones bioquímicas, funcionales o morfológicas, que se van a traducir en morbilidad e incluso mortalidad. De esta definición se destacan varios hechos: la naturaleza química de los tóxicos, la relación dosis-tiempo-respuesta y la toxicidad selectiva. Los venenos se definen de manera semejante, sólo que su origen es botánico o a partir de las secreciones de ciertos animales.
En la historia de la toxicología moderna son tres los personajes que han dado las bases para su desarrollo como ciencia: Paracelso, Claude Bernard y Orfila. Theophrastus Bombastus von Hohenheim, quien escogió el pseudónimo de Paracelso, fue quien en la Edad Media estableció el concepto fundamental de la dosis en toxicología para explicar por qué una misma sustancia podía causar o no daño, o lo que era más importante, actuar como remedio o como veneno. Lo anterior lo resumió en su famoso apotegma: "dosis sola facit venenum ", lo que se podría traducir como que todo es veneno dependiendo de la dosis.
Ya en el siglo XIX destacan el francés Claude Bernard y el español nacionalizado francés José Mateo Buenaventura Orfila; el primero introduce el método científico en toxicología al emplear diversos venenos, el curare por ejemplo, para el estudio de la neurofisiología. Orfila, a quienes muchos llaman el "Padre de la Toxicología Moderna", estudia los aspectos patológicos y forenses de las intoxicaciones, y retomando los conceptos de Paracelso, establece la necesidad no sólo de identificar a los tóxicos, sino de cuantificarlos en los especímenes biológicos para de esta manera establecer la relación entre la dosis y el efecto. Las implicaciones de este concepto en la clínica y en la solución de problemas médico-legales tienen vigencia hasta la actualidad.
El mismo Orfila reconoció que si bien la toxicología en sus principios emanó de la farmacología, en el transcurso de los siglos adquirió características propias para ser una ciencia independiente. A la luz de los conocimientos actuales se reconoce el genio visionario de Orfila ya que la toxicología moderna no se limita únicamente al estudio de los efectos adversos causados por los medicamentos, sino que se extiende al estudio de otros numerosos grupos de agentes químicos como son los plaguicidas, los disolventes orgánicos, los metales y metaloides, los humos, vapores y gases, y los plásticos, amén de los numerosos venenos botánicos y animales.
De cualquier manera, se destaca que entre las diversas ciencias en que se apoya la toxicología, la farmacología ocupa un lugar preponderante pues de ella toma muchos de sus modelos para explicar tanto el curso temporal de los tóxicos en el organismo, como sus mecanismos de acción.
Otras disciplinas relacionadas con la toxicología son la bioquímica, la fisiología, la patología, la inmunología, la medicina forense, la salud pública, la epidemiología y la ecología.
Tomando los modelos farmacológicos, se puede estudiar cómo actúa el organismo sobre el tóxico y la toxicodinamia, que explica como actúa el tóxico sobre el organismo.
El movimiento de los tóxicos en el organismo o toxicocinética, se puede estudiar siguiendo la mnemotecnia ADME, que es la sigla de absorción, distribución, metabolismo y excreción (Figura 1).
Figura 1
Resumen de la Toxicocinética y la Toxicodinamia de los compuestos químicos en el organismo. (ADME, Absorción, Distribución, Metabolismo, Eliminación.)
La vía gastrointestinal es con mucho la más importante en toxicología; la absorción de un compuesto y, por ende, su biodisponibilidad, está sujeta a múltiples factores dependientes del tóxico (dosis, concentración, forma física, propiedades fisicoquímicas) y del individuo (edad, presencia de alimento, pH, motilidad, reabsorción enterohepática, y otros). La superficie total disponible para absorción por esta vía es cercana a los 200 m2 y la distancia que tiene que recorrer la molécula tóxica para alcanzar la circulación es de aproximadamente 40 micrómetros (µm).
La vía inhalatoria es la entrada de agentes químicos presentes en el aire en forma de humos, vapores, gases, neblina o rocíos.
La superficie de absorción alveolar es también alrededor de 200 m2 pero la distancia que recorre el tóxico para llegar a los capilares es muy corta, de 1 a 2 µm; esta es la vía de absorción más importante en toxicología industrial.
La piel es un órgano de contacto directo con los múltiples tóxicos presentes en el ambiente contra los que actúa como una barrera eficiente. Su superficie de absorción es inferior a las antes mencionadas, de 1.7 m2 para un adulto de 70 kg y la distancia de translocación es superior a los 100 µm. Hay, sin embargo, algunas excepciones de importancia en toxicología clínica; así, algunos agentes como los disolventes orgánicos, el tetraetilo de plomo, las anilinas y los insecticidas organofosforados, pueden absorberse rápidamente por la piel a través de los folículos pilosebáceos, pudiendo causar casi inmediatamente manifestaciones de intoxicación aguda.
Relacionados con el proceso de absorción, en particular con la vía digestiva, se debe mencionar el llamado efecto de primer paso y de la circulación enterohepática. El efecto de primer paso se refiere a la proporción de la dosis que puede ser eliminada por el hígado, el intestino u otros sitios de biotransformación, antes de su llegada a la circulación general. La circulación enterohepática adquiere particular importancia en toxicología y da las bases para aplicar algunos procedimientos para prevenir la reabsorción intestinal de los tóxicos. Se sabe que una vez que estos compuestos llegan al hígado sufren biotransformación en metabolitos que pueden ser inertes o igual o más tóxicos que el compuesto original (el paratión se biotransforma en paraoxón que es cincuenta veces más tóxico que el primero). Estos últimos se eliminan a través de la bilis y llegan al intestino, en donde son reabsorbidos, y al llegar a la circulación general van a seguir ejerciendo sus efectos tóxicos.
La administración de dosis múltiples de carbón activado mediante el procedimiento conocido como diálisis gastrointestinal, permite adsorber tanto al compuesto original, como a sus metabolitos activos, bloqueando así su reabsorción y por lo tanto su circulación enterohepática.
Una vez alcanzada la circulación general se inicia el proceso de distribución a todo el organismo. En este fenómeno se involucran diversos factores como el pH sanguíneo, la unión de la molécula tóxica a las proteínas plasmáticas y a los tejidos, la perfusión en estos mismos, la hidro- o lipofilicidad. Destaca la unión a las proteínas, principalmente la albúmina y en segundo lugar las globulinas. Una cantidad del compuesto tóxico no se une a estos elementos constituyendo la llamada "fracción libre" en contraposición a la "fracción unida”. Las alteraciones de las proteínas plasmáticas como la hipoalbuminemia (hepatopatías, nefropatías, desnutrición o la hipoalbuminemia transitoria de la menstruación), dan lugar a un incremento de la fracción libre y, por ende, la posibilidad de causar efectos tóxicos.
Un concepto farmacológico teórico, el llamado volumen de distribución aparente (Vda), es de gran importancia en toxicología para aplicar ciertos procedimientos tendientes a incrementar la excreción de tóxicos. Por definición el Vda es el volumen de los diferentes compartimientos en los cuales se distribuye un compuesto en la misma concentración que alcanzó en el compartimiento plasmático. Cuando esta distribución es en los compartimientos acuosos, el Vda es por lo general 1/kg (100%); cuando la distribución ocurre en otro tipo de compartimientos, en particular los ricos en tejido graso, el Vda tiende a ser muy elevado, en ocasiones superior a 10 a 15 U/kg (1 000 a 1 500%).
Ejemplos de medicamentos con sus respectivos Vda se muestran en el Cuadro 1. Como regla general, aquellos agentes con Vda 1/kg, son susceptibles de ser removidos de los tejidos y fácilmente eliminados con el uso de diuréticos y algunos procedimientos dialíticos, no así en aquellos con Vda > 1/kg, en los cuales estos procedimientos no son útiles. En general aquellos compuestos que se unen en forma extensa a las proteínas plasmáticas tienen un Vda pequeño, mientras que en aquellos que tienen una unión baja a las proteínas, sus Vda son elevados.
| Cuadro 1. Volúmenes de distribución de algunos compuestos de interés toxicológico | ||
| Agente | Volumen de distribución (Vd = L/kg) |
Equivalencia en porciento |
| Aciclovir | 0.69 | 69% |
| Ácido acetilsalicílico (aspirina) | 0.15 | 15 |
| Amitriptilina | 14.0 | 1 400 |
| Anfotericina B | 4.0 | 400 |
| Cafeína | 0.61 | 61 |
| Carbamazepina | 1.4 | 140 |
| Cimetidina | 1.0 | 100 |
| Clonazepam | 3.2 | 320 |
| Clorpromazina | 21.0 | 2 100 |
| Cocaína | 2.1 | 210 |
| Diazepam | 1.1 | 110 |
| Digoxina | 7.0 | 700 |
| Etanol | 0.54 | 54 |
| Flunitrazepam | 3.3 | 330 |
| Haloperidol | 17.8 | 1 780 |
| Imipramina | 23.0 | 2 300 |
| Meperidina | 4.4 | 440 |
| Naproxeno | 0.16 | 16 |
| Nicotina | 2.6 | 260 |
| Nortriptilina | 18.0 | 1 800 |
| Paracetamol (acetaminofén) | 0.95 | 95 |
| Ranitidina | 1.8 | 180 |
| Teofilina | 0.5 | 50 |
| Trimetoprim | 1.8 | 180 |
| Valproico, ácido | 0.13 | 13 |
| Warfarínicos (1ª generación) | 0.11 | 11 |
| Warfarínicos (2ª generación) | 1.0 | 100 |
El nombre de metabolismo es incorrecto para esta fase de la toxicocinética, pero se ha conservado para facilitar el recuerdo del acrónimo ADME; lo correcto en todo caso es biotransformación. La biotransformación se considera un mecanismo clave de defensa del organismo, mediante el cual las reacciones químicas transforman los compuestos xenobióticos en el cuerpo.1
Ya se adelantó al referirse a la biotransformación, que la principal vía de excreción o eliminación de químicos y metabolitos hacia el exterior del organismo es la renal, pero también son de utilidad la intestinal, a través de la exhalación pulmonar, el sudor, la leche materna y otras más.
Estudia la interacción entre las moléculas de los tóxicos y los sitios específicos de acción, los receptores. La unión del tóxico (T) con el receptor (R), da lugar a una nueva molécula a partir de la cual se origina el estímulo que produce el efecto tóxico en el órgano o célula:
T + R = TR - Estímulo - Efecto
La toxicodinamia es compleja y variada, por lo que su descripción va más allá de los fines de esta Lección. Sólo se hará mención somera de algunos mecanismos a manera de ejemplo.
Es con mucho uno de los mecanismos más frecuentes de toxicidad. Tanto los xenobióticos como sus metabolitos pueden inhibir ciertas enzimas en su sitio de acción. La inhibición puede ser irreversible o reversible. Un ejemplo de este mecanismo de toxicidad es la intoxicación causada por los insecticidas orgánico-fosforados. Estos compuestos inhiben específicamente la acetilcolinesterasa a nivel de las hendiduras sinápticas, uniéndose en forma covalente a su sitio estérico. Lo anterior trae como consecuencia que la acetilcolina no se desdoble en acetato y colina y, por lo tanto, estimula en forma continua a las células efectoras: músculo liso, músculo estriado, glándulas y neuronas, lo que origina las manifestaciones clínicas de la intoxicación por estos compuestos.
Un ejemplo más: muchos metales actúan también por este mecanismo debido a la afinidad de determinadas enzimas por los sulfhidrilos (-SH) en el sitio activo. El plomo, por este mecanismo, inhibe varios pasos de la vía sintética del grupo prostético heme provocando alteraciones en la formación de la hemoglobina.
La molécula del tóxico guarda relación con los sustratos para ciertas conversiones metabólicas, siendo así aceptada por las enzimas en uno o más de los pasos de la cadena bioquímica, dando lugar a la síntesis de un producto anormal altamente tóxico. Un ejemplo es el fluoroacetato de sodio que por este mecanismo inhibe algunos pasos del ciclo de Krebs.
Muchos metales actúan como cofactores en reacciones enzimáticas. Algunos agentes quelantes remueven estos metales alterando la respuesta enzimática. El ditiocarbamato guarda relación con el disulfiram y tiene la capacidad de quelar los iones de cobre de la enzima deshidrogenasa aldehídica que interviene en la biotransformación del etanol. La ingestión de éste simultáneamente con el ditiocarbamato, aun en mínimas cantidades, da lugar a un “efecto antabuse” característico.
El ácido cianhídrico (HCN), al unirse selectivamente con el hierro de los sistemas citocromooxidasa, hace perder la capacidad de oxidación de este último, bloqueando la respiración aeróbica e impidiendo así el paso de oxígeno al interior de las células. El resultado final es asfixia bioquímica que puede originar la muerte en pocos minutos.
Otros mecanismos de asfixia bioquímica son la formación de carboxihemoglobina por acción del monóxido de carbono, de metahemoglobina por acción del monóxido de carbono, y de metahemoglobina secundaria a intoxicación por diversos agentes oxidantes: dapsona, anilinas, nitrobenceno y otros.
Diversos fármacos interfieren en la neurotransmisión del impulso nervioso (neurolépticos, antidepresores tricíclicos, curare, nicotina, insecticidas orgánico-fosforados, peyote, LSD). Otros se unen con las macromoléculas celulares dando lugar a necrosis, por ejemplo, el metabolito activo causa necrosis hepática. Al interferir en la síntesis de los ácidos nucleicos, algunos compuestos causan mutagénesis, embriogénesis y carcinogénesis.
En fin, y de gran actualidad, se han identificado numerosos agentes como productores de radicales libres responsables de la toxicidad aguda o tardía como en el caso del paraquat, el monóxido de carbono y los insecticidas orgánico-fosforados cuyos síndromes tardíos difieren sustancialmente de las manifestaciones agudas. Numerosos compuestos químicos alteran en distinto grado la respuesta inmune dando lugar al desarrollo de una nueva disciplina: la inmunotoxicología.
Las intoxicaciones accidentales o intencionales integran un motivo frecuente de consulta en los servicios de urgencias e incluso en consultorios médicos, siendo de vital importancia la evaluación inicial, la identificación del tóxico, el diagnóstico y el tratamiento oportuno, con la finalidad de evitar complicaciones y la mortalidad. En efecto, la intoxicación puede producir un amplio espectro de síntomas, signos o hallazgos clínicos, por esta razón es indispensable que el médico de primer contacto integre un diagnóstico certero.
De ahí, que se conozca el término síndrome tóxico o “toxíndrome” que hace referencia al conjunto de signos y síntomas que se presentan tras la exposición a una sustancia tóxica. A continuación, se describen dichos toxíndromes.
Se produce por un aumento de la actividad simpática, secundario al consumo excesivo de cocaína, anfetaminas, efedrina, pseudoefedrina, metilxantinas (teofilina, aminofilina), cafeína y fenilpropanolamina. Clínicamente se presenta con hipertensión arterial, taquicardia, taquipnea, agitación, alucinaciones, paranoia, ansiedad, midriasis, hipertermia, diaforesis, e incluso convulsiones.2
Existe una variedad importante de fármacos con propiedades anticolinérgicas, los cuales inhiben competitivamente la unión del neurotransmisor acetilcolina a los receptores muscarínicos (de acetilcolina), dichos receptores se ubican en sistema nervioso central, corazón, nervios periféricos del músculo liso intestinal y bronquial además de glándulas secretoras (salival y sudoríparas) y cuerpo ciliar del ojo.3
Cuando se presenta una intoxicación por anticolinérgicos los agentes más comúnmente involucrados son: antihistamínicos, antidepresivos tricíclicos, ciclobenzaprina, agentes antiparkinsonianos, antiespasmódicos, fenotiazinas, atropina, escopolamina y alcaloides de la belladona. El cuadro clínico se caracteriza por ansiedad, agitación, desorientación, alucinaciones, delirio, paranoia coma, mioclonías, hipertermia, taquicardia, taquipnea, hipertensión arterial, midriasis, piel seca y enrojecida, mucosas secas, disminución de ruidos intestinales y retención urinaria.3
Este síndrome se presenta tras el consumo de sustancias descritas como “alucinógenas” entre las que se encuentran el LSD, la fenciclidina, mescalina, psilocibina, anfetaminas como el MDMA (éxtasis) y dextrometorfano; su principal efecto es la alteración de la percepción sensorial, del pensamiento y del estado de ánimo, se observa agitación, midriasis, nistagmo, hipertermia, sinestesias y taquicardia. Habría que decir que también puede presentarse un síndrome serotoninérgico, representado por la alteración en el estado mental, anormalidades neuromusculares y manifestaciones autónomas.4
En relación con los opioides, éstos se consideran sustancias naturales y sintéticas que poseen una actividad semejante a la morfina, por consiguiente, existen fármacos que se recetan ampliamente para manejo del dolor, entre ellos la morfina y la buprenorfina, sin embargo, debido a los efectos de estos compuestos se han usado de forma recreativa, fomentando así el abuso de su consumo.5
La intoxicación por opioides se caracteriza por una tríada clásica: miosis puntiforme, coma y depresión respiratoria, también se presenta bradipnea, bradicardia, hipotensión arterial, edema pulmonar, hipotermia, peristalsis intestinal disminuida e hiporreflexia.5
La etiología más frecuente de este síndrome es por abuso en el consumo de benzodiazepinas, pero también se encuentran implicados otros fármacos como los barbitúricos, relajantes musculares (carisoprodol), alcoholes y zolpidem. Se manifiesta clínicamente por depresión del sistema nervioso central, confusión, estupor, coma, miosis, hiporreflexia pupilar, hipotermia, bradicardia, bradipnea o apnea, hipotensión arterial y disminución de reflejos osteotendinosos.6
Los plaguicidas de tipo organofosforados y carbamatos actúan como potentes inhibidores de la enzima acetilcolinesterasa lo que les confiere la capacidad de causar toxicidad colinérgica grave tras su exposición ya sea inhalada, ingerida o por contacto cutáneo. Dentro de la etiología de este síndrome también se encuentran los medicamentos que poseen mecanismo de acción similar como fisostigmina, edrofonio, betanecol y urecolina, y agonistas colinérgicos como la pilocarpina. Los signos y síntomas que se presentan son confusión, coma, miosis, bradicardia, hipertensión o hipotensión arterial, taquipnea o bradipnea, salivación, incontinencia urinaria y fecal, diarrea, vómito, diaforesis, epífora, espasmos gastrointestinales, broncoconstricción, convulsiones, fasciculaciones, mioclonías y debilidad muscular.7
La serotonina es un neurotransmisor que se encarga de la modulación de la atención, el comportamiento y la termorregulación, existen fármacos cuyos mecanismos de acción aumentan la neurotransmisión serotoninérgica, entre éstos se unbican los inhibidores de la recaptura de serotonina, los inhibidores de la monoaminooxidasa, meperidina, dextrometorfano y L-triptófano. El síndrome serotoninérgico ocurre cuando existe la combinación de estos fármacos o por sobreingesta de los mismos.8
Clínicamente se caracteriza por confusión, agitación, delirio hiperactivo, coma, midriasis, hipertermia, taquicardia, taquipnea, hipertensión arterial, temblor, mioclonías, hiperreflexia osteotendinosa, clonus, trismo, rigidez muscular, diaforesis, rubor y diarrea.8
El tratamiento general de las intoxicaciones se lleva a cabo en tres fases consecutivas principales: el manejo de la emergencia en el sitio donde ocurrió la intoxicación (fase de emergencia), la cual se basa en retirar al paciente de la fuente de exposición, se inicia la descontaminación y las maniobras de reanimación básica;9 la aplicación de medidas generales, sintomáticas y de sostén para salvar la vida del paciente, mantener estables sus signos vitales y corregir las complicaciones que se presenten (fase de apoyo vital avanzado); finalmente, el tratamiento de detoxificación, general y específico tendiente a prevenir la absorción del tóxico, a incrementar su excreción y su neutralización mediante el empleo de antídotos y antagonistas (fase de detoxificación o descontaminación).
Para los fines de esta Lección, se hará la descripción de los principales fármacos empleados para el tratamiento específico de algunas intoxicaciones. Cabe destacar que se trata de medicamentos en general muy eficaces, aunque desafortunadamente su número es limitado por lo que, salvo algunas excepciones, no sustituyen a las dos fases de emergencia y de apoyo vital.
Un antídoto es un químico capaz de combinarse con un tóxico para dar lugar a un nuevo compuesto, en general inerte, polar y fácilmente eliminable. Un antagonista está relacionado estructuralmente con el tóxico que actúa como agonista, y compite con éste por el sitio específico de su receptor modificando así la respuesta. En casos particulares, el fármaco puede actuar por otros mecanismos que no son los mencionados. A continuación, se describirán los principales agentes empleados con estos fines.
El carbón activado se obtiene de la pirólisis de diversos materiales orgánicos como madera, pulpa, hueso, almidón, lactosa, sacarosa y cáscara de coco.
Se "activa" sometiendo el carbón a corrientes de aire o vapor de agua calentadas a temperaturas elevadas (600-900 °C), con lo que se fragmentan los gránulos de carbón incrementando así su superficie de adsorción que para el carbón activado disponible es de 950 m2/g; asimismo existen productos considerados “súper activados” cuya superficie promedio es de 800 a 3 500 m2/g lo que se traduce en mayor adsorción de las sustancias.10
A través de fuerzas de unión de Van der Waals pueden unirse a una gran cantidad de moléculas químicas que al ser adsorbidas se inactivan y se eliminan unidas al carbón. Por muchos años se utilizó el carbón activado en dosis única después del lavado gástrico, extrayéndolo a través de la misma sonda con los remanentes del tóxico. En años recientes ha ocurrido un cambio trascendente en la forma de utilizar este agente, lo que ha llevado a considerar que se trata del redescubrimiento de un compuesto secular que cumple con el sueño de los toxicólogos: contar con un antídoto casi universal.
El término diálisis gastrointestinal se acuñó al comparar el efecto del carbón activado con la administración de albúmina durante la diálisis peritoneal, con lo que se incrementaba de manera importante la excreción de los tóxicos. El procedimiento ha demostrado ser eficaz en la depuración no renal de numerosos tóxicos y sus metabolitos en particular cuando siguen circulación enterohepática, misma que al ser bloqueada evita su reabsorción y por lo tanto que continúen ejerciendo sus efectos adversos.
El método se aplica a múltiples medicamentos, compuestos químicos diversos, hongos y plantas venenosas. En el Cuadro 2 se anotan ejemplos de estos agentes tóxicos en los cuales la diálisis gastrointestinal ha mostrado su utilidad. Por el contario, existen xenobióticos que poseen pobre absorción con el carbón activado, como son: álcalis, clorpropamida, doxepina, etanol y otros alcoholes, los metales pesados, imipramina, hierro, litio, metotrexato, potasio, tobramicina, valproato de sodio y la vancomicina.11
| Cuadro 2. Ejemplos de tóxicos que se depuran eficazmente mediante diálisis gastrointestinal con dosis repetidas de carbón activado11 | |
| Fármacos | Paracetamol, antiinflamatorios no esteroides, aspirina (y otros salicilatos), benzodiazepinas, carbamazepina, cloroquina, clorpropamida, ciclosporina, dapsona, dextropropoxifeno, digitoxina, digoxina, difenhidramina, difenilhidantoína, efedrina, fenilbutazona, fenilpropanolamina, fenobarbital, gentamicina, imipramina (y otros antidepresores tricíclicos), metamizol (dipirona), metotrexato, nadolol, piroxicam, pseudoefedrina, propranolol, quinina, teofilina, tobramicina, valproato y vancomicina |
| Químicos | Insecticidas orgánico-fosforados, paraquat, raticidas warfarínicos, fosfato de zinc, alfa-cloralosa, sulfato de talio |
| Hongos | Amanita vera, A. virosa, A. phalloides |
| Otros | Diversas plantas venenosas |
La técnica es la siguiente:
Desde 1985, la Food and Drug Administration (FDA) de Estados Unidos aprobó a la N-acetilcisteína como antídoto de la sobredosis por paracetamol (acetaminofén) para prevenir el desarrollo de necrosis hepática fulminante que caracteriza a la intoxicación por este medicamento.
El empleo de la N-acetilcisteína como antídoto preventivo, se fundamenta en el conocimiento de la farmacología del paracetamol y las alteraciones que causa en casos de sobredosis (Figura 2).
Figura 2
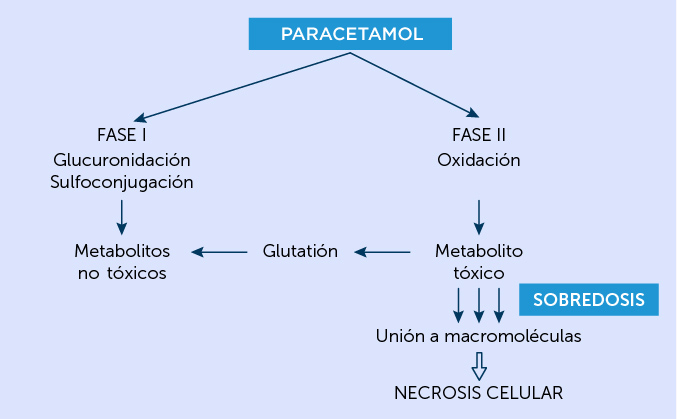
Biotransformación del paracetamol (acetaminofén) y mecanismos de toxicidad.
En dosis terapéuticas el paracetamol se biotransforma en el hígado mediante la conjugación con ácido glucurónico (40 a 67%) y sulfato (20 a 46%), en metabolitos atóxicos; al mismo tiempo, una pequeña parte (5 a 15%) resulta oxidada por el citocromo P450, la cual se convierte en N-acetil-p-benzoquinoneimina, que en presencia de glutatión se transforma en conjugados de mercaptato o cisteína, cabe señalar que ambos conjugados son inocuos y rápidamente eliminados por vía urinaria. Ahora bien, en casos de sobredosificación de paracetamol, no hay suficiente glutatión hepático, lo que produce un daño tisular directo por el acúmulo excesivo de N-acetil-p-benzoquinoneimina libre.12
El resultado de esta alteración se manifiesta inicialmente como toxicidad en la zona hepática tipo III (centrolobulillar) y en casos graves puede afectar también a las zonas I y II, causando destrucción total del parénquima hepático.12
La administración de N-acetilcisteína da lugar a la formación de cisteína, precursora del glutatión, con eso se incrementan los niveles depletados de este último conjugándose el metabolito activo libre para así prevenir la necrosis hepática. Los mejores resultados se obtienen cuando la N-acetilcisteína se administra en las siguientes 8 a 10 h12 que siguen a la sobredosis de paracetamol lo que en general previene la necrosis. Hasta las 24 h12 contribuye a disminuir la mortalidad por esta intoxicación aun en el 50%, después de este tiempo su utilidad es mínima y la mortalidad se eleva a 80 al 100% de los casos.
La N-acetilcisteína se puede administrar por vía oral y por vía intravenosa, siendo esta última la de primera elección, aunque es importante señalar que presenta riesgo de reacciones alérgicas (muy escaso). Para su administración se usarán viales de 2 a 6 g al 20% disueltos en solución glucosada al 5%; la dosis inicial es de 150 mg/kg en 200 mL de solución glucosas al 5% en adultos y de 3 mL/kg de solución glucosada al 5% en niños, para pasar en 45 min. Se continuará con 50 mg/kg en 500 mL de solución glucosada al 5% para adultos y 10 mL/kg de solución glucosada al 5% en niños para 4 h. Finalmente, se administrarán 50 mg/kg en 500 mL de solución glucosada al 5% para adultos y 20 mL/kg de solución glucosada al 5% en niños para 16 horas.12
El antídoto en general es bien tolerado y ocasionalmente se han descrito algunos efectos secundarios moderados como eritema cutáneo transitorio o gastroenteritis, que no son indicaciones para suspenderlo. Aunque en un principio fue motivo de controversia, en el momento actual se considera útil su administración aun si hay manifestaciones de encefalopatía. Es importante tener en mente que, dada la gravedad de esta intoxicación, en casos de duda es preferible administrar el antídoto.
En el momento actual, además de ser considerado el antídoto de elección en la intoxicación por paracetamol, la N-acetilcisteína se ha empleado al parecer con resultados prometedores en el tratamiento de otras intoxicaciones en donde se involucran la producción de radicales libres o metabolitos reactivos: cloroformo, tetracloruro de carbono, 1,2-dicloropropano, acrilonitrilo, doxorrubicina, ciclofosfamida y paraquat. Asimismo, se recomienda para la prevención de los síndromes tardíos en las intoxicaciones por monóxido de carbono e insecticidas orgánico-fosforados.
La atropina se utiliza para el tratamiento inicial de la intoxicación por insecticidas orgánico-fosforados y carbamatos. Corrige fundamentalmente las manifestaciones muscarínicas, pero al proteger a los receptores de la acción de la acetilcolinesterasa, da tiempo suficiente para que actúen los antídotos reactivadores de esta enzima y así tratar íntegramente estas intoxicaciones. Es muy importante recordar que para que la atropina actúe como un antídoto, es necesario que se administre en dosis elevadas, esto es, hay que intoxicar al paciente con atropina para salvarlo de la intoxicación por los agentes anticolinesterásicos.
Se administra por vía endovenosa, ya sea en forma de "bolos" o por infusión continua, a la dosis de 2 a 5 mg en adultos, cada 3 a 5 min y de 0.05 mg/kg cada 5 a 10 min en niños, según necesidad, duplicando la dosis previa en caso de no haber presentado mejoría. Se debe mantener la atropinización administrando dosis repetidas conforme sea necesario por 2 a 12 h basado en la persistencia o recurrencia de los síntomas.13 Una vez que se alcanza la atropinización (disminución de secreciones, sequedad de piel y mucosas, midriasis, taquicardia), la atropina se administrará por razón necesaria en caso de que reaparecieran manifestaciones muscarínicas.
El azul de metileno es el cloruro de tetrametiltionina, este compuesto actúa aceptando un electrón de NADPH (Nicotinamida-adenina dinucleótido fosfato) y de reductasa de la hemoglobina, convirtiéndose a azul de leucometileno. Éste a su vez es capaz de reducir la metahemoglobina a hemoglobina (convierte el hierro férrico a ferroso). El azul de metileno es el tratamiento de elección para las metahemoglobinemias tóxicas cuya etiología incluye numerosos oxidantes directos e indirectos: anilinas, acetofenetidina, nitritos, nitratos, cloratos, nitrobenceno, benzocaína, lidocaína, nitroglicerina, nitroprusiato, fenazopiridina, sulfonamidas, clorobenceno, naftaleno, nitrofenol, nitrato de plata y trinitrotolueno; aunque excepcional, el mismo azul de metileno en dosis elevadas produce metahemoglobinemia. En el momento actual los agentes que con mayor frecuencia están produciendo metahemoglobinemias tóxicas son la benzocaína y la lidocaína.
El azul de metileno se presenta en ampollas de 10 mL con 100 mg del fármaco (10 mg/mL). Se administra diluido en 30 a 50 mL de solución glucosada al 5%, no se recomienda utilizar solución de NaCl al 0.9% porque reduce la solubilidad del azul de metileno; la dosis inicial es de 1 a 2 mg/kg, por infusión endovenosa a pasar en 3 a 5 min. Si el caso lo requiere se puede repetir la dosis en 30 a 60 min, tomando en cuenta que los niveles tóxicos de azul de metileno se pueden presentar en dosis totales de 7 mg/kg.
En el caso, por demás raro, de metahemoglobinemia secundaria a sobredosis de azul de metileno, es de utilidad la administración endovenosa de ácido ascórbico o una exanguinotransfusión urgente..
En la actualidad se considera como un antídoto verdadero en la intoxicación causada por los antidepresores tricíclicos y sustituye a la fisostigmina que no ha demostrado efectividad, además de poseer efectos indeseables importantes.
El bicarbonato de sodio actúa como antídoto favoreciendo la unión de la fracción libre de los tóxicos a las proteínas plasmáticas con lo que se reduce la concentración de dicha fracción y, por lo tanto, sus efectos dañinos, sobre todo las alteraciones del ritmo cardiaco y de la tensión arterial.
En años recientes, se ha introducido el uso de bicarbonato de sodio administrado por nebulizaciones, para el tratamiento de las exposiciones agudas, generalmente graves, debido a la inhalación de gases o vapores irritantes como el cloruro o el amoniaco. Los criterios de administración del bicarbonato en intoxicación por antidepresivos tricíclicos son: complejo QRS superior a 0.11 s, presencia de arritmias ventriculares o hipotensión grave.12
Además, continúa vigente su empleo para alcalinizar la orina y de esta manera evitar la reabsorción de fármacos con pH ácido como la aspirina y el fenobarbital.
La dosis de bicarbonato de sodio en las intoxicaciones sistémicas o para alcalinizar la orina son de 1 a 2 mEq/kg/dosis, IV, cada 4 a 6 h. Para neutralizar los efectos de los vapores o gases irritantes, se disuelven 3 mL de una solución de bicarbonato de sodio al 8.4% en 2 mL de solución salina, con lo que se obtiene una solución al 5%, misma que se administra mediante nebulización.
Las sales de calcio, en particular el cloruro y el gluconato, tienen aplicación en diversas emergencias toxicológicas: sobredosis de medicamentos bloqueadores de los canales del calcio, etilenglicol, fluoruros, magnesio y envenenamiento por picadura de viuda negra (o Lactrodectus mactans).
Estos fármacos se presentan para su administración endovenosa, únicamente: el cloruro de calcio al 10% contiene 1.36 mEq de Ca, en tanto que el gluconato contiene 0.45 mEq del ion. Ambos se administran lentamente por la vía mencionada; el cloruro a la dosis de 0.2 mL/kg/dosis, en tanto que el segundo a la dosis de 0.6 mL/kg/dosis. Es importante el monitoreo electrocardiográfico durante esta terapéutica, así como las determinaciones de calcio sérico, total y ionizado, para prevenir la hipercalciemia iatrogénica.
Como antídoto del hierro se administran 20 mg/kg/dosis hasta una dosis máxima de 80 mg/kg/día o 6 g/día; se administra14 por vía endovenosa, cada 4 a 6 h. Es importante señalar que la deferoxamina vía oral podría no tener un efecto benéfico, por el contrario, puede existir el riesgo de aumentar la sideremia cuando se usa de este modo, debido a que podría verse facilitada la absorción intestinal de la ferrioxamina.14
De manera característica el quelado resultante, al ser eliminado por la orina, le confiere a ésta un color rojizo, mismo que puede ser utilizado como indicador del tiempo que debe administrarse la deferoxamina. Cuando los niveles de hierro han descendido, el color de la orina retorna paulatinamente a la normalidad; cuando esto ocurre es el mejor signo para suspender la administración de la deferoxamina.
Algunos medicamentos del tipo de los neurolépticos, como el haloperidol y en particular la metoclopramida, muestran, en casos de sobredosis, una toxicidad selectiva por los núcleos grises basales en donde inhiben los receptores dopaminérgicos, dando lugar a la acción predominante del otro neurotransmisor que mantiene el equilibrio funcional de estos núcleos, la acetilcolina. El resultado son las manifestaciones clínicas de "extrapiramidalismo": acatisia, crisis bucolinguales y oculógiras, distonías de torsión de cuello y tronco, y en pacientes mayores, pseudoparkinsonismo.
Para corregir esta intoxicación se han utilizado numerosos medicamentos, siendo la difenhidramina el único que ha demostrado su efectividad. Su relación estructural con la atropina y la benzatropina y su afinidad por los núcleos grises, le permiten revertir los efectos resultantes de la acción de la acetilcolina. Inicialmente se administra por vía endovenosa lenta a la dosis de 1 a 2 mg/kg/dosis que se puede repetir a las 4 h. Una vez que las manifestaciones clínicas han remitido, es conveniente continuar administrando la difenhidramina por la vía oral por un mínimo de 72 h, con lo que se previenen las recaídas de la intoxicación que es resultado del incremento de la vida media de los fármacos ocasionado por la sobredosis.
Agente quelante sintetizado en 1940 y empleado desde la década de 1960 como antídoto de metales pesados; sin embargo, es hasta años recientes en que ha ocurrido su introducción progresiva en la clínica. Estructuralmente relacionado con el BAL (anti-Lewisita británica),15 no posee los efectos secundarios indeseables de éste y tiene además la ventaja de su administración por vía oral. Sus principales indicaciones son las intoxicaciones causadas por el mercurio, el arsénico y el plomo (en este último caso puede aplicarse succimer solo o en combinación con el ácido etilendiaminotetraacético).15 La dosis es de 10 mg/kg/dosis, cada 8 h por los primeros cinco días, continuando a la misma dosis pero cada 12 h por 14 días más.
En cuanto al dimercaprol o anti-Lewisita británica, éste se administra por vía intramuscular profunda; en caso de intoxicación severa por arsénico u oro, la dosis es de 3.5 a 5 mg/kg cada 4 h por 6 dosis, posteriormente cada 6 h por 4 dosis, y cada 8 h por 3 dosis, seguido de cada 12 h por dos dosis, y finalmente una vez al día durante 10 días. Para intoxicación por mercurio la dosis inicial es de 5 mg/kg cada 4 h por 1 a 2 días continuando con 2.5 mg/kg cada 12 ó 24 h durante 10 días. En el caso de intoxicación por plomo, la dosis es de 4 mg/kg cada 4 h por 3 días comenzado la quelación con EDTA con una segunda dosis, seguido de 2.5 mg/kg durante 1 a 4 días.15
No obstante que se ha preconizado su seguridad en comparación con otros quelantes, el DMSA ocasionalmente puede causar efectos secundarios que deben tenerse en mente: náusea, vómito, diarrea, meteorismo, eritema, prurito, rinorrea, mareos, parestesias, eosinofilia, trombocitosis y elevación transitoria de las transaminasas. Es posible además la movilización y excreción de algunos elementos esenciales como el zinc y el cobre.
Es el antídoto para las intoxicaciones por metanol y etilenglicol. Actúa inhibiendo la biotransformación del primero en formaldehído y del segundo en glicoaldehído, metabolitos ambos responsables de los efectos dañinos de estos alcoholes.
Si se tuviera a la mano una forma farmacéutica de etanol estéril se puede administrar por vía endovenosa; en caso contrario, es preferible emplear la vía oral para lo cual se le administra directamente diluido en jugos o a través de una sonda nasogástrica. Para la vía endovenosa se recomienda una solución de etanol al 10%, para la oral al 20% (esto se consigue fácilmente diluyendo bebidas que contienen etanol al 40% ―ginebra, vodka― en cantidad similar de jugo). Iniciar con una dosis de impregnación de 600 a 80016 mg/kg en 30 a 60 min, y de sostén de 100 mg/kg/h, con lo que se consigue una concentración sanguínea de etanol de 100 mg/dL.
El flumazenil es un antagonista competitivo de los receptores de las benzodiazepinas con mínimas propiedades agonistas. Se conocen bien su farmacocinética y su farmacodinamia, en las que destacan su baja unión a las proteínas del plasma (54 al 64%) y su vida media corta (t½: 53 min). Experimentalmente se ha visto que la administración de 1.5 mg de flumazenil lleva a bloquear el 55% de los receptores, en tanto que con 15 mg la ocupación de los mismos es total.
Se administra por vía endovenosa y para el caso se han recomendado diversos regímenes terapéuticos, de los que el más útil es el siguiente: administrar inicialmente 0.2 mg por vía intravenosa durante 30 s, pudiéndose administrar dosis repetidas de 0.2 mg hasta alcanzar una dosis máxima de 1 mg y hasta lograr el efecto deseado. En niños, la dosis inicial es de 0.01 mg/kg por vía intravenosa durante 15 s, con una dosis máxima de 0.2 mg.6
El inicio de acción del flumazenil se presenta en aproximadamente 1 a 2 min, con una respuesta del 80% en los primeros 3 minutos.15
En años previos, se sugirió el uso de flumazenil como parte del tratamiento en la encefalopatía hepática; sin embargo, existe evidencia de baja calidad que sugiere un efecto beneficioso a corto plazo del flumazenil sobre este padecimiento en personas con cirrosis hepática, por lo que se requiere de pruebas adicionales de alta calidad que pueden evaluar los posibles beneficios y daños de este fármaco en personas con diagnóstico de encefalopatía hepática.17
El glucagón es un polipéptido secretado por las células alfa del páncreas. En el pasado se le utilizó como tratamiento inicial de pacientes en coma después de sobredosis y los resultados obtenidos hicieron que esta práctica fuera abandonada.
El glucagón se considera un antídoto de primera línea en la sobredosis por betabloqueadores, el cual se considera efectivo en el inicio del tratamiento, aunque puede volverse ineficaz en el manejo prolongado debido a la presencia de taquifilaxia.18
Se administra por vía intravenosa, como una dosis de bolo lenta seguida de una infusión continua. La dosis inicial es de 5 mg para pasar en un minuto, en caso de que no aumente el pulso o la presión arterial después de 10 a 15 min, se deberá administrar un segundo bolo, posteriormente al aumento de la presión arterial o del pulso, se iniciará una infusión a una velocidad de 2 a 5 mg/h. En población pediátrica, la dosis inicial es de 50 µg/kg, debe observarse un efecto dentro de los primeros 3 min con una respuesta máxima a los 5 a 7 min; si después de ese tiempo no se observa ningún efecto, se administra una segunda dosis. El objetivo del tratamiento es mantener una presión arterial media de 60 mm Hg.18
Sin lugar a dudas es uno de los antagonistas más utilizados en toxicología. Es un antagonista competitivo de alta afinidad de los receptores opioides que actúa desplazando las moléculas de estos fármacos de su sitio de unión (receptores mu, kappa, sigma), resultando en una inhibición de sus efectos.19
La intoxicación aguda por opioides se manifiesta clínicamente por la tríada clásica representada por: depresión del estado de consciencia, miosis y depresión respiratoria, sin embargo, existen otras características clínicas especificas de cada agente, por ejemplo, la metadona, que además causa prolongación del intervalo QT y torsade de pointes. Debido a lo anterior, el tratamiento inicial debe centrarse en el soporte de las vías respiratorias con la posterior administración de naloxona, preferiblemente por vía intravenosa con la finalidad de revertir los signos clínicos de intoxicación por opioides.20,21
La dosis dependerá del estado clínico debido a que, en pacientes que presenten apnea, la dosis inicial de naloxona será de 0.2 a 1 mg; en pacientes en paro cardiorrespiratorio será de 2 mg como mínimo, y en aquellos que presenten ventilaciones espontáneas se administrarán 0.4 a 0.5 mg con posterior ajuste de la dosis hasta que la frecuencia respiratoria sea de 12 o más respiraciones por minuto. La dosis deberá repetirse cada 2 a 3 min, y en caso de que no se presente respuesta satisfactoria después de 10 mg, se deberá considerar un diagnóstico alternativo. El objetivo de la administración de naloxona no es un nivel normal de consciencia, sino una ventilación adecuada.20
Después de restaurar la ventilación, pueden requerirse dosis repetidas de naloxona, por lo que se recomienda preparar una infusión, tomando en cuenta la dosis inicial total requerida para restablecer la respiración y se administrarán dos tercios de esa dosis cada hora.20
Si se retrasa la obtención de un acceso intravenoso, la naloxona puede ser administrada por vía nasal, subcutánea o intramuscular, tomando en cuenta que estas rutas son de absorción más lenta y poseen una eliminación tardía.20
En general, la naloxona es un fármaco bien tolerado, excepcionalmente se ha informado de casos aislados con reacciones adversas incluyendo hiper- o hipotensión, disritmias cardiacas y edema pulmonar.
Tales efectos se han observado en pacientes que han recibido anestesia general y en los que se han empleado múltiples medicamentos, o bien, además de los efectos depresores de los opiáceos, el paciente evoluciona con alteraciones cardiovasculares como choque cardiogénico o secuelas de disfunción cardiorrespiratoria.
Las oximas constituyen un grupo de fármacos de los cuales los más utilizados en la clínica son la obidoxima y la pralidoxima (toxogonin y 2 PAM, respectivamente). A nivel farmacológico, ambos compuestos actúan como reactivadores de la acetilcolinesterasa inhibida por la acción de los numerosos insecticidas organicofosforados: paratión, malatión, coumafos, diclorvos, dimetoato, fentión, otión, mipafux, nalex, fosfamidón, y otros.
Dado que su concentración plasmática máxima ocurre en 2 a 3 h, la administración de las oximas debe ser precedida de la "atropinización" del paciente, misma que protege inmediatamente a las células efectoras estimuladas por la acetilcolina. Su vida media en pacientes intoxicados es cercana a las 4 h; se debe administrar por vía endovenosa, en el caso de la pralidoxima la dosis de carga es de 2 g durante 20 min seguido de 0.5 g/h durante un máximo de 7 días o hasta que no se requiera de atropina.22 Con respecto a la obidoxima la dosis es de 250 mg por vía endovenosa en aquellos pacientes con predominio de afectación del sistema nervioso central, requiriendo de repetirse la dosis a las 2 y 4 h por vía oral, hasta un máximo de 5 mg/kg.15,22
Es importante recordar que estos fármacos deben administrarse lentamente por infusión endovenosa, ya que la administración rápida puede causar espasmo laríngeo o disfunción cardiorrespiratoria súbita.
Estos fármacos no deben emplearse en las intoxicaciones por carbamatos puesto que estudios llevados a cabo con uno de estos agentes, el carbaril, han mostrado que el uso de oximas empeora la intoxicación, al parecer porque el complejo formado (carbamato-oxima) puede inhibir de manera más potente a la acetilcolinesterasa que el carbaril solo.
Agente quelante obtenido por hidrólisis de la penicilina, de la que conserva sus propiedades antigénicas, se ha empleado con éxito para el tratamiento de la intoxicación por talio, plomo, cobre, mercurio y arsénico. Se utiliza por la vía oral a la dosis de 30 a 50 mg/kg/día, en dos tomas durante diez días. De acuerdo con la eliminación de los metales y la evolución clínica de los pacientes, se pueden repetir otras series del quelante.
Cuando se emplea como antídoto su única contraindicación es la alergia probada a la penicilina; por lo demás es en general bien tolerado y con mínimos efectos secundarios.
Cuando se administra como tratamiento de enfermedades crónicas del tejido conectivo como la artritis reumatoide, el empleo prolongado de la D-penicilina puede originar efectos indeseables graves como síndrome nefrótico, neuritis óptica y lupus farmacoinducido. Esto no se ha observado en los tratamientos cortos de las intoxicaciones por metales.
Corresponde al ácido etilendiaminotetraacético-calcicodisódico (EDTA CaNa2), agente quelante potente para administración endovenosa. Posee un amplio espectro para quelar metales pero se ha utilizado principalmente en el tratamiento de las intoxicaciones por plomo, zinc, manganeso, cobre, mercurio, cadmio y berilio.
Se administra a la dosis de 20 a 30 mg/kg/dosis, por infusión endovenosa, a pasar en 2 h. Repetir cada 24 h por periodos de cinco días. En la encefalopatía plúmbica se recomienda administrar el versenato combinado con la D-penicilamina o el DMSA. El empleo de dosis altas puede causar nefrotoxicidad tubular y glomerular. Algunos efectos secundarios, en general raros y transitorios, son malestar general, fatiga, fiebre, lagrimación y congestión nasal, hipotensión y depleción de otros metales.
Es el antídoto para contrarrestar los efectos de los anticoagulantes warfarínicos. En casos de ingestión o inhalación particularmente de rodenticidas warfarínicos de primera o segunda generación, aun en ausencia de sangrados, de manera preventiva se administra la vitamina K por vía intramuscular a las dosis de 0.5 a 1.0 mg/dosis en lactantes, 1.0 a 2.0 mg/dosis en escolares y de 5 a 10 mg en adolescentes y adultos.
Los warfarínicos de segunda generación son muy potentes y pueden causar sangrados extensos que ponen en peligro la vida; en estos casos la vitamina K debe administrarse por vía endovenosa, a dosis de 10 mg en infusión lenta (20 a 60 min),23 debiendo vigilar estrechamente a los pacientes ante la posibilidad de que desarrollen reacciones de hipersensibilidad.
En estos pacientes, una vez controlada la urgencia debe continuar administrándose vitamina K1 (fitonadiona) por la vía oral, a la dosis de 2 a 5 mg/día, por el tiempo necesario hasta que se recuperen los tiempos de protrombina y parcial de tromboplastina. Esto puede tardar meses en el caso específico de los warfarínicos de segunda generación, que en general poseen vidas medias de eliminación muy prolongadas.
El oxígeno en concentraciones de 100% es el antídoto de la intoxicación por monóxido de carbono. La hipoglucemia es una causa común del agravamiento de pacientes intoxicados por etanol, salicilatos, hipoglucemiantes orales y hierro, misma que revierte rápidamente con el empleo de dextrosa. Si se utiliza la dextrosa hipertónica (50%), vigilar estrechamente al paciente para evitar el efecto contrario, ya que la hiperglucemia puede a su vez causar efectos adversos graves. La protamina está indicada en la intoxicación por heparina; 1 mg neutraliza 100 mg de la heparina.
El tratamiento antituberculoso con isoniazida puede originar como efecto adverso algo similar a la deficiencia de vitamina B6, cuya principal manifestación son convulsiones, mismas que se corrigen con la administración de piridoxina. El clorhidrato de tiamina es un tratamiento coadyuvante útil en la intoxicación etílica, ya que contribuye a corregir varios efectos adversos resultantes de la dificultad de entrada del piruvato al ciclo del ácido tricarboxílico; la tiamina precisamente facilita esta entrada incrementando así la producción de ATP.
Los fragmentos de anticuerpos específicos de digoxina, se han utilizado en el tratamiento de la intoxicación por este último glucósido. Estos fragmentos comúnmente referidos como Fab, actúan uniéndose a la fracción libre intravascular de la digoxina responsable de sus efectos tóxicos, permitiendo su difusión al espacio intersticial y de ahí a su eliminación urinaria. Este antídoto es efectivo, pero desafortunadamente su precio es muy elevado y no está disponible en México. Una alternativa muy útil para esta intoxicación es la diálisis gastrointestinal con dosis múltiples de carbón activado.
El 4-metil pirazol está siendo estudiado como un antídoto coadyuvante del etanol, para el tratamiento de las intoxicaciones causadas por el metanol y el etilenglicol. Administrado por la vía oral, al parecer actúa como un potente inhibidor de la biotransformación hepática del metanol y del etilenglicol con lo que se previene la formación de los metabolitos responsables de los efectos tóxicos. Falta mayor experiencia, pero promete ser un antídoto muy eficaz.
Para finalizar, se destaca que de algunos agentes que por muchos decenios se emplearon para el tratamiento de intoxicaciones, en el momento actual hay muchas dudas acerca de su eficacia, amén de poseer numerosos efectos secundarios indeseables; tal es el caso de la ipecacuana, de la fisostigmina y del monoacetato de glicerilo.