Figura 12
Etiología de hipotiroidismo congénito primario.
Coordinador: Dr. Miguel Ángel Rodríguez Weber
Hipotiroidismo congénito
Dra. María de la Luz Ruiz Reyes, Dr. Raúl Calzada León
El hipotiroidismo congénito (HC) es la enfermedad endocrina más frecuente en la etapa neonatal. Es indispensable su detección y tratamiento oportuno en esa etapa para evitar alteraciones graves en el desarrollo cerebral que conducirían a un retraso mental frecuentemente profundo e irreversible, ya que las hormonas tiroideas son imprescindibles para la formación, migración e interconexión de las neuronas tanto en la etapa prenatal como en la posnatal y son indispensables para regular diversas funciones metabólicas.
Hipotiroidismo congénito se refiere a la disminución de la síntesis y/o acción de las hormonas tiroideas desde el nacimiento. Puede ser causado por una producción deficiente de hormonas tiroideas o, menos frecuentemente, por una resistencia a su acción en los tejidos diana.
La prevalencia de hipotiroidismo varía según el lugar geográfico y la población. En nuestro país afecta a uno de cada 2 426 recién nacidos vivos, y es más frecuente en las mujeres en una relación de 2 a 1 con respecto a los varones.
Anatómicamente, el hipotiroidismo se clasifica en primario, cuando existe un defecto de la glándula tiroides, o central, debido a la falta de estimulación tiroidea por la hormona estimulante de la tiroides (TSH) como resultado de una patología hipofisiaria (secundario) o por la falta de la hormona liberadora de tirotropina (TRH) por alguna afección hipotalámica (terciario).
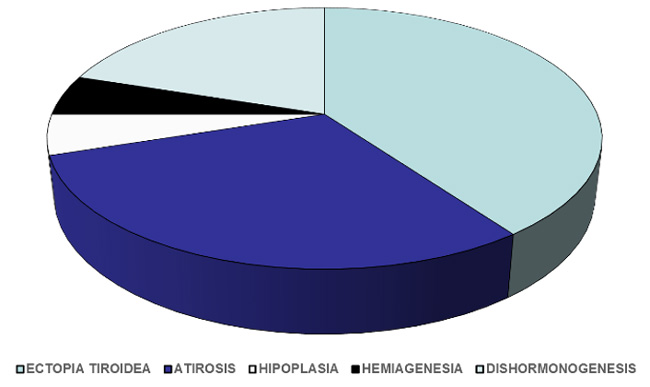
Es el más común, tradicionalmente es subdividido en disgenesia tiroidea, que se refiere a un espectro de anormalidades del desarrollo tiroideo que ocurre en aproximadamente 95% de los casos, y en dishormonogénesis, en donde se encuentra alterada la síntesis de hormonas tiroideas en pacientes con una glándula tiroides estructuralmente intacta y en localización habitual.
La disgenesia tiroidea se origina cuando la glándula tiroides no concluye el desarrollo y no finaliza la migración como en el nódulo tiroideo o ectopia tiroidea, o bien, porque no se desarrolló tejido glandular, lo que se conoce como agenesia tiroidea o atirosis. En muy pocos casos las causas son la hipoplasia tiroidea o la hemiagenesia tiroidea. Una causa de atirosis iatrogénica es cuando la madre con hipertiroidismo recibe tratamiento con I131 durante la etapa de embriogénesis tiroidea del producto.
La mayor proporción de pacientes con disgenesia tiroidea se presentan como una entidad esporádica. Las causas genéticas implican genes que median la diferenciación, la migración y el crecimiento de la tiroides, sin embargo, menos del 5% de los casos son atribuibles a una mutación. En contraste, la mayoría de los individuos con dishormonogénesis presentan mutaciones en genes que codifican componentes conocidos en la biosíntesis de la hormona tiroidea: NIS/SLC5A5, PDS/SLC26A4, TPO, Tg, DUOX2, DUOXA2 y la yodotirosina desyodasa IYD. Algunos estudios sugieren que más del 50% de los pacientes con tiroides eutópica (anatómicamente normal y en su sitio), pueden tener mutaciones en uno y a menudo en dos o más de estos genes (Cuadro 16).
| Cuadro 16. Síndromes asociados a disgenesia tiroidea | |
| Gen | Características clínicas |
| PAX8 | Alteraciones urogenitales |
| NKX2.1 | Enfermedad pulmonar intersticial, corea |
| FOXE1 | Sx de Bamforth-Lazarus: paladar hendido, úvula bífida, atresia de coanas, cabello erizado |
| NKX2.5 | Cardiopatías congénitas |
| JAG1 | Sx Alagille: cardiopatía congénita y defectos variables en cara, ojos, hígado y esqueleto |
| GLIS3 | DM neonatal, retraso del desarrollo, glaucoma congénito, fibrosis hepática, riñones poliquísticos |
Se consideraba poco frecuente e inicialmente se estimaba su incidencia en 1:29.00 a 1:110.00 recién nacidos, sin embargo, los datos actuales en los Países Bajos, los que utilizan una rigurosa estrategia de tamiz neonatal, demuestran que la incidencia en HCC puede ser tan alta como 1:16 000.
Se produce por el desarrollo anormal del hipotálamo y/o la hipófisis, que aunque puede afectar la producción de TSH en forma aislada, en 75% de los pacientes produce deficiencia combinada de hormonas hipofisiarias: TSH, GH, Prolactina, ACTH, LH, FSH.
Algunos de estos casos son secundarios a mutaciones genéticas en factores de transcripción, involucrados en el desarrollo y crecimiento del hipotálamo y de la hipófisis, HESX1, LHX3, LHX4, OTX2, SOX3, PRP1 y POU1F1 que ocasionan hipoplasia o agenesia hipofisiaria.
Rara vez el HCC puede ser causado por defectos genéticos específicos en la señal de TRH o de TSH, la más común de éstas, es una mutación inactivante recientemente descrita en el gen IGSF1, contenido en el cromosoma X, que codifica una glucoproteína en la superficie celular que parece promover la expresión normal del receptor de TRH en los tirotropos. Otras causas muy raras, son mutaciones en el receptor de TRH, en la subunidad β de TSH y en TBL1X, que es otro gen ligado al cromosoma X.
Los pacientes con HCC con deficiencia hipofisiaria combinada, pueden diagnosticarse clínicamente por micropene (hipogonadismo hipogonadotrópico), hipoglucemia o ictericia neonatal prolongada (insuficiencia suprarrenal), o falta de crecimiento posnatal (deficiencia de hormona de crecimiento). Frecuentemente en estos pacientes podemos observar alteraciones en la línea media: paladar alto o hendido, úvula bífida o incisivo central único, criptorquidia bilateral y micropene.
Algunos pacientes con una tiroides estructural y funcionalmente normal, presentan una incapacidad temporal para sintetizar y secretar hormonas tiroideas. Esto sucede en prematuros, en la deficiencia o exceso de yodo, antecedentes de uso de medios de contraste yodados, medicamentos antitiroideos, anticuerpos maternos que atraviesan la barrera placentaria, deficiencia de selenio, mutaciones heterocigotas de DUOX2 o DUOXA2, hemangiomas hepáticos con niveles elevados de desyodasa tipo 3, o por ajustes en la función tiroidea que se tienen que producir en la etapa fetal o neonatal ante la existencia de una enfermedad grave que afecta de manera importante el balance nutricional y/o funcional. Cuando hay un mecanismo de resolución espontánea y autolimitado, este efecto es transitorio y la función tiroidea se normaliza (Figura 12).
Figura 12
Etiología de hipotiroidismo congénito primario.
La glándula tiroides se deriva del endodermo del piso de la faringe, que en la tercera semana de gestación inicia la diferenciación de las células foliculares. A partir del día 32 se forman los lóbulos tiroideos cuyas células proliferan hasta la 8ª semana, al mismo tiempo que se observa migración desde el piso del estomodeo hasta situarse por debajo del cartílago cricoides. Aunque la diferenciación de las células foliculares termina en la 10ª semana, no es hasta la 12ª semana que estas células expresan el receptor para TSH con lo que inician la captura de yodo para sintetizar T3, T4 y tiroglobulina. Sin embargo, la regulación del eje hipotálamo-hipófisis- tiroides se completa hasta la semana 20 de gestación. Por lo tanto, en las primeras 20 semanas de gestación (sdg) 100% de los requerimientos fetales de hormonas tiroideas se obtienen a través del paso transplacentario de hormonas tiroideas maternas y no es hasta las 38 sdg que el producto es capaz de sintetizar 100% de sus requerimientos hormonales.
Si la madre presenta hipotiroidismo gestacional y no es tratada de manera adecuada, o bien cuando existe una disfunción placentaria que afecte el aporte de hormonas tiroideas de la madre hacia el producto, éste presentará una deficiencia de hormonas tiroideas que ocasionarán trastornos irreversibles a nivel de sistema nervioso central, aunque el feto tenga una tiroides con estructura y función normales.
Por otro lado, cuando el producto tiene hipotiroidismo congénito, y esto se asocia a la falta de aporte materno durante la gestación, presentará trastornos graves en el desarrollo del sistema nervioso central y retraso de la maduración esquelética que pueden identificarse desde el nacimiento, lo cual se traduce en un mayor riesgo de afección del neurodesarrollo.
A nivel hipotalámico, en el núcleo paraventricular, se sintetiza la TRH, hormona reguladora de la síntesis y secreción de TSH en la hipófisis. La unión de TSH a su receptor, regula la yodación y la síntesis hormonal de la célula folicular en la glándula tiroides.
La hormona predominantemente producida por la tiroides es la T4, que es una prehormona que se convierte en tejidos periféricos a la hormona biológicamente activa: T3, la cual tiene hasta 15 veces mayor afinidad al receptor de TSH que la T4. La unión de la TSH a su receptor folicular, estimula la síntesis de hormonas tiroideas y el crecimiento de la glándula tiroides, la circulación de hormonas tiroideas inhibe la secreción de TRH y TSH en el hipotálamo y la hipófisis, completando el mecanismo de retroalimentación negativo, que mantiene así la homeostasis tiroidea.
El yodo, por lo tanto, es un elemento necesario para la síntesis de las hormonas tiroideas. Se obtiene a través de la dieta y lo ideal es ingerir 150 µg de yodo diario. En las mujeres embarazadas los requerimientos aumentan a 200 µg/día. La deficiencia de yodo da origen a trastornos en el crecimiento y en el desarrollo cerebral, que se manifiestan por bocio y cretinismo.
La mayoría de los pacientes presentan signos y síntomas tan leves que suelen pasar desapercibidos, y cuando se acentúan las manifestaciones y permiten su identificación, es frecuente que ya exista daño neurológico (Cuadro 17).
| Cuadro 17. Manifestaciones clínicas en hipotiroidismo congénito | |
| Fontanela anterior amplia | Hipoactividad |
| Fontanela posterior > 0.5 cm | Anemia, palidez |
| Llanto ronco | Hipotonía |
| Hernia umbilical | Alteración en la deglución |
| Succión débil | Reflujo gastroesofágico |
| Ictericia prolongada | Facies tosca |
| Piel seca | Crecimiento lento de uñas |
| Intolerancia al frío o hipotermia | Maduración ósea retrasada |
| Macroglosia | Somnolencia |
Los niños con HC presentan una mayor frecuencia de malformaciones asociadas: cardiopatías, nefropatías, gastrointestinales y esqueléticas, por lo que se deben descartar durante su evolución una vez confirmado el diagnóstico.
La detección oportuna de hipotiroidismo congénito debe ser una prioridad en todo el mundo. Uno de los avances más importantes en el diagnóstico temprano de esta patología es la realización de tamiz neonatal en recién nacidos que se inició en Quebec y Pittsburg desde 1974, y en nuestro país desde 1988 con la expedición de la Norma Técnica 321 y posteriormente en la Norma Oficial Mexicana NOM 007-SSA2-1993, la cual establece su realización como una acción obligatoria en todo el personal involucrado en la atención maternoinfantil. De esta manera, se ha logrado identificar a la población de riesgo y después, al confirmar diagnóstico, se les indica el tratamiento oportunamente para evitar el retraso mental de los pacientes.
Se basa en la determinación de TSH y/o T4 total en papel filtro con características especiales (tarjeta de Güthrie) en el que se vierten gotas de sangre obtenida por punción del talón de los recién nacidos entre el tercer y el quinto día de vida. Mediante este estudio, es factible detectar los casos de hipotiroidismo congénito primario, en los cuales el nivel de TSH estará elevado. La muestra de cordón umbilical no se recomienda debido a una alta frecuencia de falsos positivos, ya que la TSH se encuentra fisiológicamente elevada al momento del nacimiento. Cuando además se determina T4 en el tamiz, se pueden detectar también los casos de HCC y aquellos con elevación tardía de TSH. En algunos lugares se está implementando determinación alternativa de TBG (globulina transportadora de hormonas tiroideas) y/o T4 libre en lugar de T4 total, aumentando la tasa de diagnóstico de hipotiroidismo central.
La ficha de identificación del tamiz neonatal debe llenarse completamente y con letra legible ya que es la única forma de localizar oportunamente los casos sospechosos de hipotiroidismo congénito. Debe incluir el nombre completo de la madre del recién nacido, teléfono y domicilio completo con detalles para su posible localización (recuerde que no es infrecuente en nuestro país que la madre pase la cuarentena en casa de la abuela), fecha del nacimiento del niño, edad gestacional, peso y talla del recién nacido, así como la fecha y sitio en donde fue tomada la muestra. La toma adecuada de la muestra en cantidad y calidad es indispensable para procesarla y, por lo tanto, una muestra inadecuada provoca retrasos innecesarios en el diagnóstico y la demora en el tratamiento oportuno. Idealmente los resultados de tamiz neonatal deben entregarse por escrito de manera individual a los padres y el pediatra debe anexar una copia a su expediente.
Cuando el reporte de tamiz muestra una TSH superior a 10 mUI/mL debe considerarse sospechoso para hipotiroidismo congénito y tenemos que localizar inmediatamente al paciente a fin de realizarle el estudio confirmatorio. Todo resultado anormal en el tamiz neonatal debe ser confirmado mediante la determinación inmediata de un perfil tiroideo que en los casos de HC primario presentará bajas concentraciones de T4 total y T4 libre en conjunto con una persistente elevación de TSH. Idealmente se debe determinar la concentración sérica de tiroglobulina en el perfil tiroideo confirmatorio. Esta glucoproteína se sintetiza exclusivamente en el tejido tiroideo, por lo que su concentración es proporcional a la cantidad de tejido tiroideo funcional.
En los casos con HC central, los valores de T4 total y T4 libre estarán bajos con niveles de TSH normal o baja.
Si los niveles de T4 total están bajos sin elevación de TSH y T4 libre normal, se debe sospechar una deficiencia de TBG, que es un trastorno recesivo ligado al X y no requiere tratamiento.
A los recién nacidos que no se les haya realizado tamiz neonatal, se recomienda que en su primera consulta de niño sano se solicite una determinación de TSH, T4 y T4 libre.
Una vez confirmado el diagnóstico, es ideal investigar la causa, y ya que las disgenesias tiroideas son causa en más de 90% de los casos, se debe investigar la localización y características de la glándula tiroides. El gammagrama tiroideo con tecnecio-99 es el estándar de oro para establecer el diagnóstico etiológico y debe realizarse antes del inicio de tratamiento. Si ya se inició el tratamiento sustitutivo con hormonas tiroideas no es recomendable realizar gammagrama tiroideo, ya que por el medicamento el tejido tiroideo se encontraría bloqueado y no se captaría en el estudio.
La ecografía tiroidea es útil y complementaria; sin embargo, se requiere un transductor especial para partes pequeñas y un radiólogo con experiencia.
La radiografía AP de ambas rodillas sirve para evaluar la madurez esquelética o edad ósea en el paciente. Debido a que las hormonas tiroideas son necesarias para madurar el sistema esquelético, en algunos casos, e independientemente de la etiología, cuando la madre fue capaz de proporcionar a través de la placenta suficiente cantidad de hormonas tiroideas hasta el momento del nacimiento, la edad ósea coincide con la edad cronológica, en tanto que cuando el aporte transplacentario fue insuficiente, la maduración esquelética es menor a la esperada para la edad gestacional. En niños nacidos a término, se espera observar el núcleo distal de fémur y el proximal de la tibia, ambos de por lo menos 1 cm de diámetro (de acuerdo al método de Pyle), por lo que cuando estos núcleos se observan se asume que el paso transplacentario de hormonas tiroideas maternas fue normal durante la vida intrauterina y que, por ende, protegió el desarrollo neurológico. Esta afección se conoce como hipotiroidismo congénito de inicio extrauterino. Por el contrario, cuando estos núcleos no están presentes o son menores al diámetro esperado en un recién nacido a término, se catalogan con una edad ósea retrasada o hipotiroidismo congénito de inicio intrauterino, lo que nos traduce un aporte materno insuficiente de hormonas tiroideas transgestacional con un alto riesgo de retraso en el desarrollo neurológico.
El nacimiento prematuro plantea problemas para el diagnóstico de HC, especialmente en aquellos con muy bajo peso al nacer en los cuales la disfunción de la tiroides incluirá hipotiroxinemia transitoria del prematuro (T4 baja con TSH normal).
Por otra parte, la maduración retardada del eje hipotálamo-hipófisis-tiroides en bebés prematuros también puede resultar en una TSH inicialmente normal en un paciente con hipotiroidismo primario pero que después se elevaría. Por lo anterior, en prematuros con resultado del tamiz normal, se sugiere realizar determinación sérica de TSH y T4 entre los 21 y los 30 días de vida para así poder diagnosticar a los pacientes con HC y resultado falso negativo del tamiz en este grupo de edad.
El reemplazo hormonal es una prioridad en estos pacientes, ya que debe instalarse en forma oportuna (a más tardar en la segunda semana de vida) para que sea factible evitar retraso mental e irreversible. La realización de estudios complementarios no debe retrasar el inicio de tratamiento ya que cada día de retraso disminuye el coeficiente intelectual del paciente. La dosis recomendada de inicio de levotiroxina es de 12 a 15 µg/kg/día, vía oral, en ayuno y cada 24 h. La levotiroxina en México se encuentra sólo en tabletas, ya que no es estable en solución o suspensión. Actualmente hay tabletas en dosis pediátricas para administrar la cantidad lo más exacta que requiera el paciente cada 24 h. Se recomienda colocar la dosis indicada en una cuchara y diluirla con agua para obtener una mezcla o suspensión que puede ser administrada y deglutida inmediatamente por el recién nacido. Una vez administrada la dosis, se debe agregar más agua a la cuchara para evitar que queden residuos de la dosis calculada en la cuchara.
Para evitar interferencias con la absorción del medicamento, se recomienda diferir la ingesta de alimentos 30-40 min posterior a su administración. Es importante referir al paciente con el endocrinólogo pediatra, quien debe evaluar todos los riesgos asociados al tipo de hipotiroidismo congénito: atresia de coanas, miotonías, problemas degenerativos o desmielinizantes, cardiopatía y otras malformaciones congénitas, alteraciones en el mecanismo de la deglución, reflujo gastroesofágico, retraso en la migración y maduración neuronal, ritmos lentos y de bajo voltaje en el mapeo cerebral, apneas o hipopneas, alteraciones en la osificación del oído medio con dificultades auditivas, problemas cerebelosos con trastornos del equilibrio, alteraciones en la adquisición y maduración de las funciones motoras finas, dificultades en la audiopercepción y visopercepción, trastornos del lenguaje, trastornos de la lectoescritura, etcétera, además de ajustar e individualizar la dosis de manera periódica y vigilar la progresión del crecimiento y el desarrollo, para así cumplir con los objetivos clínicos y bioquímicos del manejo integral del paciente con hipotiroidismo congénito.
Es necesario el monitoreo de TSH y T4L de forma regular durante los primeros cuatro años de vida, para asegurar que la dosis es adecuada, que los niveles de laboratorio se encuentran en los valores esperados para la edad, para evaluar la evolución del crecimiento, logros en el neurodesarrollo y para corroborar una adecuada adherencia del tratamiento.
Además del tratamiento sustitutivo y por el riesgo de retraso en el neurodesarrollo inminente, los pacientes con hipotiroidismo congénito requieren de forma obligatoria un programa de estimulación integral de neurodesarrollo, que inicie desde el momento de la confirmación del diagnóstico, donde se capacitará y supervisará a los padres en los procesos secuenciales del desarrollo y les otorgará herramientas y recursos para el aprendizaje, así como las estrategias para la rehabilitación, lo que en conjunto permitirá recuperar y conseguir un desarrollo organizado y avanzar en su maduración evitando secuelas neurológicas.
Cuando se inició el tratamiento con levotiroxina ante la imposibilidad de confirmar el diagnóstico, debe continuarse el tratamiento y monitoreo hasta los tres años de edad neurológica, es decir, cuando el paciente logre subir y bajar escaleras sin apoyar las manos y alternando los pies y además tenga control diurno y nocturno del esfínter vesical y anal. Cuando se logra esta edad madurativa, es factible suspender el tratamiento por cuatro a seis semanas para realizar perfil tiroideo, y gammagrama tiroideo, para así poder confirmar o descartar el diagnóstico sin lesionar el neurodesarrollo. Actualmente debe considerarse la evaluación con estudios moleculares en algunos pacientes, de acuerdo a sus manifestaciones clínicas, lo que permite establecer la asociación entre alteraciones genéticas, trastornos tiroideos y el patrón de herencia.