Figura 1
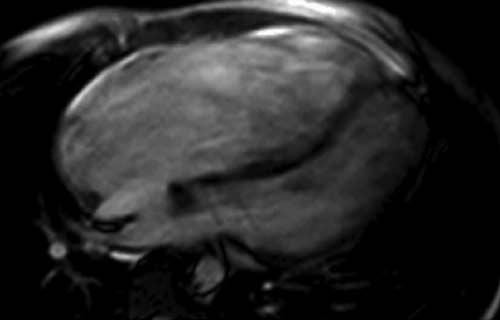
Resonancia magnética de anomalía de Ebstein con un gran adosamiento que condiciona que la parte ventricular funcional sea mínima. Se aprecia el abombamiento septal hacia el ventrículo izquierdo.
Coordinador: Dr. Juan Verdejo Paris
Aspectos de interés en la cardiopatía congénita del adulto
Dr. Juan Calderón-Colmenero
Las cardiopatías congénitas fueron definidas por Mitchell et al., como una anomalía estructural evidente del corazón o de los grandes vasos intratorácicos con una repercusión real o potencial.1-3 Se considera que son las malformaciones más frecuentes al nacer y la prevalencia reportada es variable pero hay un consenso de que el promedio mundial va de 6 a 8 por 1 000 recién nacidos vivos.1 Al relacionar la prevalencia con el número de nacimientos por año en nuestro país (alrededor de 2 000 000), se puede inferir que cada año nacen alrededor de 12 a 16 mil niños con algún tipo de malformación cardiaca. A nivel mundial, se estima que anualmente nacen alrededor de 1 300 000 niños con una cardiopatía congénita significativa y que alrededor de 50% mueren por infección o insuficiencia cardiaca, sobre todo en los primeros años de la vida y se considera que sólo entre 2 y 15% de los pacientes son llevados a intervenciones curativas. Existe un contraste marcado entre los niños con cardiopatía congénita (CC) que nacen en los países de más altos ingresos en los cuales 85% alcanzarán la edad adulta en comparación a países con menores ingresos como Sudán, donde sólo al 15% de los niños que requieren una cirugía se la practican.4
Si bien se desconoce la prevalencia de las cardiopatías congénitas en nuestro país, podemos tener una idea sobre su importancia y repercusión con sustento en las tasas de mortalidad que en 1990 la ubicaban en sexto lugar como causa de muerte en los menores de un año. Pasó al cuarto en 2002 y se constituyó en la segunda causa de mortalidad a partir del año 2005. La primera causa de mortalidad la constituyen las complicaciones del prematuro, y en lo que corresponde a los niños entre uno y cuatro años, de ser la novena causa en 1990, escaló a la tercera en 2002 y desde el 2013 es la segunda causa sólo después de los accidentes. En la investigación de Torres-Cosme et al.,5 se encontró que de 1988 a 2013 la mortalidad infantil secundaria a cardiopatías congénitas en toda la república se incrementó en 24.8%, con un aumento en el número de decesos de 114.4 a 146.4 por 100 mil recién nacidos. En el último año del estudio (2013) se documentó un total de 3 593 decesos consecutivos a cardiopatías congénitas, un tercio de ellos ocurrió durante la primera semana de vida y los factores de riesgo encontrados fueron género masculino, nacer en hospitales no institucionales y en zonas rurales. Los autores concluyeron que las cardiopatías congénitas son un serio problema de salud pública en nuestro país, por lo que es necesaria una detección temprana y un monitoreo epidemiológico.
En relación con los diferentes tipos de cardiopatía congénita se cuenta con un análisis de 2 257 pacientes realizado en un hospital de tercer nivel de la Ciudad de México en el que se observó que la persistencia del conducto arterioso representó el 20% de los casos, situación muy explicable por la altitud de la Ciudad de México y zonas conurbadas; le siguió la comunicación interauricular (17%), comunicación interventricular (11%), tetralogía de Fallot y atresia pulmonar con comunicación interventricular (9.3%), coartación aórtica y estenosis pulmonar (3.6%), respectivamente, y la conexión anómala total de venas pulmonares (3%).6
En una revisión de pacientes adultos (> 18 años) en el Instituto Nacional de Cardiología atendidos en los últimos 20 años, se encontró que la comunicación interauricular, la persistencia del conducto arterioso, la comunicación interventricular y la coartación aórtica fueron las cardiopatías más frecuentes. Es importante mencionar que al tratarse de una institución de referencia los datos que se pueden ver en el Cuadro 1 no deben tomarse como un estudio epidemiológico. También en esa institución se observó que en los pacientes que acuden a consulta externa por primera vez la cardiopatía congénita rebasa a aquellos que acuden por problemas coronarios o valvulares. En Estados Unidos se considera que existe una población adulta con cardiopatía congénita mayor de 1 millón de pacientes, de los cuales 40 a 45% necesitan seguimiento de por vida y 25% atención especializada en unidades de referencia. Por otra parte, se calcula que en México hay alrededor de 350 mil adultos con cardiopatía congénita que requieren control y se espera un incremento anual de aproximadamente 5%, dada la tendencia actual; en un futuro cercano el volumen de pacientes adultos con cardiopatía congénita será mayor que el de niños.7-11
| Cuadro 1. Cardiopatías más frecuentes en edad adulta | |
| CardiopatÍa | NÚmero |
| Comunicación interauricular | 501 |
| Persistencia del conducto arterioso | 163 |
| Coartación aórtica | 141 |
| Comunicación interventricular | 138 |
| Estenosis aórtica | 93 |
| Aorta bicúspide | 91 |
| Anomalía de Ebstein | 68 |
| Estenosis pulmonar valvular | 54 |
| Conexión anómala total de venas pulmonares | 40 |
| Tetralogía de Fallot | 39 |
En países de altos ingresos se considera que entre 10 y 15% de las cardiopatías congénitas son diagnosticadas hasta la edad adulta y casi siempre son cardiopatías de baja complejidad, no ocurre igual en países de medianos y bajos ingresos, donde los adultos con CC no han sido diagnosticados de manera oportuna o recibido un tratamiento adecuado y no es infrecuente que tengan cardiopatías complejas. En el estudio realizado por Márquez-González et al.,12 en un hospital de tercer nivel se encontró que en 30% de los casos las cardiopatías congénitas fueron diagnosticadas en la edad adulta y otro 30% si bien fueron diagnosticados en edad pediátrica no continuaron o finalizaron su tratamiento de la cardiopatía congénita.
Los adultos con cardiopatía congénita se pueden dividir en tres grupos. El primero, conformado por pacientes que han sido sometidos a algún tipo de reparación quirúrgica; el segundo por pacientes sometidos a cirugía paliativa; y el tercero por pacientes que han evolucionado sin tratamiento quirúrgico o de cateterismo intervencionista, por un diagnóstico tardío que no es infrecuente llegue a ocurrir en casos como la comunicación interauricular y la coartación aórtica. También puede suceder en pacientes con circulación sistémica y pulmonar balanceada; los pacientes con cardiopatías complejas pueden permanecer asintomáticos hasta que dicho balance se altera, al igual que otros pacientes no candidatos a algún tipo de tratamiento quirúrgico.
Como ya se comentó, en este último grupo un excelente ejemplo es la comunicación interauricular, que es la cardiopatía más frecuente en la edad adulta, y la buena tolerancia se debe a que el cortocircuito ocurre entre cámaras de baja resistencia, como es el caso de las aurículas. La evolución empieza a modificarse entre los 30 y 40 años, a consecuencia del desarrollo de hipertensión pulmonar secundaria, arritmias supraventriculares o a la asociación a una cardiopatía isquémica o hipertensiva; otro ejemplo muy significativo es la coartación aórtica, la cual es detectada al encontrar cifras arteriales elevadas, y también la aorta bivalva con obstrucción poco significativa, en la cual el pequeño gradiente entre el ventrículo izquierdo y la aorta permite una sobrevivencia sin síntomas. Sin embargo, en un porcentaje de ellos, se puede modificar al incrementar el grado de obstrucción debido a calcificación o fibrosis, o bien por un cuadro de endocarditis bacteriana.
Hay otras cardiopatías complejas, como la anomalía de Ebstein o la transposición corregida de grandes arterias, que pueden tener en ocasiones poca traducción clínica. La anomalía de Ebstein se caracteriza por un adosamiento anormal de grados variables de los velos valvulares de la tricúspide desde el anillo auriculoventricular, normalmente situado a lo largo del endocardio del ventrículo derecho. Varía desde una discreta alteración anatómica, hasta el defecto grave, que incide en forma significativa en la calidad de vida (Figura 1). Cuando el adosamiento es ligero los pacientes tienen buena capacidad funcional y mínimas alteraciones a la exploración cardiovascular. En ocasiones la malformación se manifiesta por episodios de taquicardia paroxística supraventricular, ya que se asocia hasta en 30% al síndrome de Wolff-Parkinson-White. Diversos autores han propuesto que la anomalía de Ebstein, por su afectación de los velos valvulares tricuspídeos y del miocardio del ventrículo derecho, se debe denominar como una miopatía del ventrículo derecho.
Figura 1
Resonancia magnética de anomalía de Ebstein con un gran adosamiento que condiciona que la parte ventricular funcional sea mínima. Se aprecia el abombamiento septal hacia el ventrículo izquierdo.
Por otra parte, en la transposición corregida de grandes arterias, sin defectos asociados como una comunicación interventricular, la aurícula derecha está conectada con el ventrículo izquierdo y éste a su vez con la arteria pulmonar y la aurícula izquierda con el ventrículo derecho y la aorta, lo que hace que el retorno venoso sistémico se dirija correctamente a la circulación pulmonar y el retorno venoso pulmonar a la circulación sistémica, es decir, la circulación está fisiológicamente corregida. Los pacientes con esta malformación manifiestan sintomatología cuando empieza a desfallecer el ventrículo derecho, que tiene la función de mantener la circulación sistémica o cuando desarrollan bloqueo AV completo; es digno de mencionarse que estos pacientes tienen 2% de riesgo por año de desarrollar bloqueo AV completo, por lo que un número importante de ellos puede requerir el implante de un marcapaso permanente.
Otras cardiopatías, como la atresia pulmonar con comunicación interventricular, son tratadas en algún momento de la edad pediátrica, colocando un tubo extracardiaco del ventrículo derecho al tronco de la arteria pulmonar y cerrando la comunicación interventricular (cirugía de Rastelli). Este tubo tiende a calcificarse y, por ende, reduce su área efectiva de flujo, lo que obliga a cambiar dicho tubo extracardiaco; los pacientes con un solo ventrículo funcional son llevados a una corrección de tipo univentricular conocida como cirugía de Fontan (Figura 2). Los pacientes sometidos a este tipo de cirugía requieren un seguimiento estricto y continuo de por vida.8-11
Figura 2
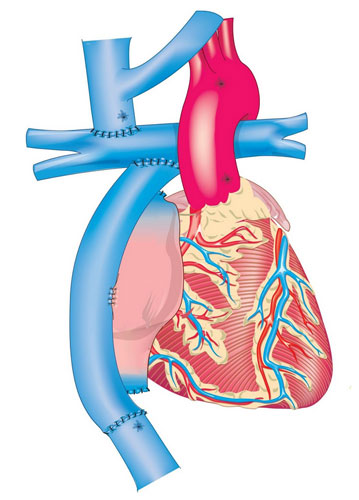
Cirugía de Fontan. Se puede apreciar el conducto extracardiaco conectando la vena cava inferior a la rama derecha de la arteria pulmonar y la anastomosis terminolateral de la vena cava superior con la misma rama. [Tomada del Banco de imágenes del Departamento de Cardiología Pediátrica del Instituto Nacional de Cardiología Ignacio Chávez.]
Otros enfermos tienen lesiones de cierto grado, que siendo candidatos a tratamiento quirúrgico, por su buena tolerancia no fueron tratados, entre ellos incluimos la comunicación interventricular de escasa repercusión hemodinámica, la comunicación interauricular, o bien pacientes con tetralogía de Fallot u otro tipo de cardiopatía compleja pero con equilibrio funcional. Por último, otras cardiopatías corregidas en la niñez permiten alcanzar la edad adulta con alguna lesión residual, lo que permite una vida aceptable o con pocas limitaciones.
La población adulta está más propensa a desarrollar arritmias, endocarditis bacteriana, descompensaciones hemodinámicas secundarias al embarazo o por la asociación de enfermedades adquiridas como la cardiopatía isquémica o la hipertensiva; este es un grupo etario en que el acceso al empleo es aún más difícil, con una cobertura limitada en los sistemas de salud o la imposibilidad de tener seguro de gastos médicos mayores, no logran casarse o tener una vida conyugal satisfactoria; estos son sólo algunos de los aspectos que se tienen que abordar de manera integral, tanto familiar como social.
Se considera que para lograr una atención óptima de los pacientes adultos con cardiopatía congénita moderada y sobre todo de gran complejidad, es indispensable una estrecha colaboración entre cardiólogos, ecocardiografistas, intervencionistas, cirujanos cardiovasculares, enfermeras y trabajadores sociales. Por lo anterior, se requiere tener en hospitales de alta especialidad un departamento dirigido al control y manejo de este tipo de pacientes. Está bien establecido que los centros con mayor volumen quirúrgico e intervencionista tienen menores índices de morbilidad y mortalidad, siendo más evidente en pacientes que requieren ser reoperados.
La evolución de los adultos con cardiopatías congénitas, con o sin tratamiento quirúrgico o intervencionista, se asocia a diversos grados de cianosis y poliglobulia, endocarditis bacteriana, arritmias supraventriculares o ventriculares y tromboembolia a nivel pulmonar o sistémico, entre otras.
La cianosis se define como la coloración azul de las mucosas, secundaria a la presencia de más de 5 g/dL de hemoglobina reducida en la sangre. La aparición de cianosis depende fundamentalmente de la concentración de hemoglobina reducida en sangre. Si la concentración es inferior a 10 g/dL, la cianosis no será detectada clínicamente, ya que con saturaciones arteriales de oxígeno de 80% o menores no se alcanzará la cantidad de hemoglobina reducida necesaria para su expresión. Existen cardiopatías congénitas con cortocircuito de sangre venosa sistémica a la circulación arterial, la magnitud de dicho cortocircuito determina la severidad de la desaturación.
En los adultos la causa más común de cianosis por cardiopatía congénita es la tetralogía de Fallot y el síndrome de Eisenmenger, en pacientes que padecen una cardiopatía congénita acianógena, llámese comunicación interventricular o persistencia del conducto arterioso, que dejadas a su historia natural desarrollan hipertensión pulmonar severa y finalmente, síndrome de Eisenmenger. La cianosis de origen cardiaco generada por cardiopatías congénitas condiciona eritrocitosis e hiperviscosidad, lo que incrementa el riesgo de tromboembolismo, trombosis in situ y riesgo de formación de abscesos cerebrales. También, en adultos con cardiopatías congénitas cianóticas hay disminución en el número de plaquetas y alteración de su función y son frecuentes las alteraciones en factores de coagulación, lo que predispone a la presentación de sangrados espontáneos o a un mayor sangrado posoperatorio.8-10
Se define a la endocarditis infecciosa (EI) como una infección endovascular de estructuras cardiovasculares (válvulas, endocardio auricular o ventricular) que incluyen endarteritis de grandes vasos torácicos (persistencia del conducto arterioso, coartación de la aorta, etc.) o cuerpos extraños intravasculares (catéteres, marcapasos, cables, desfibriladores) que se encuentren en contacto con el torrente sanguíneo. La endocarditis bacteriana es más frecuente en la edad adulta que en la pediátrica como producto de la mayor longevidad, mayor incidencia de lesiones degenerativas valvulares, así como la presencia de materiales protésicos, marcapasos, dispositivos intravasculares. La incidencia de la endocarditis bacteriana, en general, es de 3 a 10 episodios/100 000 personas-año, la cual se ha incrementado debido a una mayor supervivencia de los niños tratados con cardiopatías congénitas (Cuadro 2). Todas las anomalías congénitas de corazón, potencialmente, son un factor predisponente para el desarrollo de endocarditis bacteriana, pero hay lesiones que implican mayor riesgo, como es el caso de la comunicación interventricular.13
| Cuadro 2. Nivel de riesgo de endocarditis bacteriana |
| Riesgo elevado |
| Antecedentes de endocarditis bacteriana |
| Válvulas protésicas |
| Cardiopatía congénita compleja |
| Conductos protésicos pulmonares o sistémicos |
| Riesgo moderado |
| Prolapso mitral con insuficiencia valvular |
| Cardiopatías congénitas sin tratamiento quirúrgico |
| Con tratamiento quirúrgico intervencionista en que se haya utilizado material protésico |
| Con defectos residuales o fístula sistémica-pulmonar |
| Riesgo bajo |
| Comunicación interauricular |
| Posoperados de CIA, CIV y PCA |
| Marcapasos |
| Prolapso mitral sin insuficiencia |
| CIV, comunicación interventricular; CIA, comunicación interauricular; CAP, persistencia del conducto arterioso. |
Se debe pensar en la posibilidad de endocarditis bacteriana en todo paciente con síndrome febril con diagnóstico de cardiopatía congénita tratada o no, presencia de soplo de tipo orgánico, en que no se tenga un foco infeccioso evidente. En dichas circunstancias debe proceder a tomar hemocultivos y dado que la bacteriemia en la EI es continua, se pueden obtener en cualquier momento. Es deseable tomar tres hemocultivos, ya que de esa forma se puede detectar el microorganismo en más de 95% de los pacientes que no hayan recibido tratamiento antibiótico previo y aproximadamente en 90% de quienes han recibido tratamiento antimicrobiano en los días previos a la obtención de la muestra.
La mayoría de las endocarditis son causadas por un restringido espectro de bacterias: Streptococcus viridans, Staphylococcus aureus y Staphylococcus epidermidis representan aproximadamente el 80% de los casos de endocarditis infecciosa. Además, se debe practicar un estudio ecocardiográfico para demostrar la presencia de vegetaciones (Figura 3). La mortalidad reportada, secundaria a endocarditis bacteriana es elevada, por lo que es primordial indicar tratamiento profiláctico en procedimientos dentales invasivos, ya que es la puerta más común de origen de la bacteriemia y si bien el riesgo de bacteriemia en el parto y cesárea es bajo, también está indicada la aplicación profiláctica de antibióticos, como la amoxicilina o la cefalotina, o bien la clindamicina en caso de alergia a la penicilina.
Figura 3
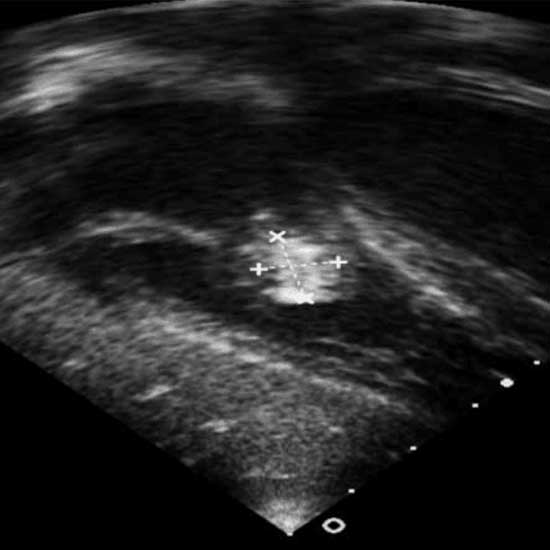
Ecocardiograma transtorácico bidimensional. Se observa una imagen de 11 mm de bordes irregulares que corresponde a una vegetación en la inserción de las valvas anterior y septal de la tricúspide que protruye hacia la cavidad ventricular.
Las arritmias son de las causas más frecuentes que descompensan al adulto con cardiopatía congénita, de ellas alrededor del 80% corresponden al flúter y a fibrilación auricular, 5 a 7% a taquicardia ventricular, disfunción del marcapaso en 5% y bradicardia entre 3 y 5%.14
Existen dos grandes categorías de arritmias en los pacientes adultos con cardiopatías congénitas. La primera se relaciona con la propia cardiopatía congénita que persiste a pesar del tratamiento quirúrgico o intervencionista. La segunda es producto del desarrollo de alteraciones electrofisiológicas secundarias a dilatación de cavidades, alteraciones hemodinámicas, hipoxemia, o bien, cicatrices quirúrgicas. En pacientes con ritmo sinusal, como sucede en pacientes con comunicación interauricular, puede haber bradicardia consecutiva a disfunción del nodo sinusal, alteración que a su vez es producida por la canulación requerida para la circulación extracorpórea.
La corrección quirúrgica del canal auriculoventricular puede condicionar alteración a nivel del nodo AV o en el tronco del haz de His, ya que el defecto septal se sitúa muy cercano al sistema de conducción. El flúter auricular es de las arritmias más frecuente en el adulto, se puede observar en la anomalía de Ebstein, el canal auriculoventricular o en pacientes sometidos a corrección de tipo univentricular (cirugía de Fontan), así como en la comunicación interauricular, tratada o no, producto de la dilatación de las aurículas u originada en la cicatriz quirúrgica. La fibrilación auricular es una arritmia que produce deterioro del estado clínico de los pacientes, por lo que debe ser tratada de inmediato para restaurar el ritmo sinusal, o por lo menos lograr una frecuencia ventricular adecuada. De no lograrse revertir, está indicado el uso de anticoagulantes, para la prevención de fenómenos tromboembólicos, así como de digital y betabloqueadores para un satisfactorio control de la frecuencia cardiaca.
Es importante resaltar las extrasístoles ventriculares en pacientes posoperados de corrección total de tetralogía de Fallot o de cirugía de Rastelli, donde se coloca un tubo extracardiaco del ventrículo derecho al tronco de la arteria pulmonar, técnica quirúrgica utilizada para la corrección de la atresia pulmonar con comunicación interventricular y el tronco arterioso común. Dichas arritmias deben ser estudiadas y tratadas porque representan un sustrato para el desarrollo de taquicardia ventricular. La muerte súbita de origen cardiaco continúa siendo un problema en los pacientes adultos con cardiopatías congénitas, ya que ocurre con mucha mayor incidencia que en la población abierta. La mayoría de las veces se presenta en pacientes portadores de tetralogía de Fallot, transposición de grandes arterias, coartación aórtica y estenosis aórtica.
La hipertensión pulmonar (HAP) se define como la presión media de la arteria pulmonar mayor de 20 mm Hg. Su etiología es múltiple, pero entre las principales causas están las cardiopatías congénitas en donde existen cortocircuitos importantes de izquierda a derecha que condicionan resistencias pulmonares elevadas con cambios en la íntima y la media de la vasculatura pulmonar, cuya gravedad puede alcanzar tal grado que se convierta en irreversible. Varios registros europeos coinciden en señalar que las CC son de las etiologías más frecuentes de HAP. Así, el registro holandés de hipertensión pulmonar pediátrica recabado entre 1991-2005 que incluyó a 3 263 pacientes, la HAP secundaria a CC representó alrededor de 80% de los casos, seguida por la HAP secundaria a problemas respiratorios con 8%. En el registro Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) de 317 pacientes que tenían HAP, el 36% se asoció a CC. En otro registro, el Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL), que incluyó a 216 pacientes menores de 18 años, la HAP secundaria a CC fue del 34.6%.15-17
Dependiendo de la edad de corrección de la CC existe un riesgo potencial de desarrollar HAP. En el registro alemán, que incluye a pacientes sometidos y no sometidos a corrección, la prevalencia de HAP varía de 3% en pacientes con persistencia del conducto arterioso a 100% en pacientes con ventana aortopulmonar.17 El riesgo de desarrollar síndrome de Eisenmenger varía según el tipo de cardiopatía; en pacientes con comunicación interauricular no corregida es de 10-17%, se eleva a 50% en pacientes con comunicación interventricular y alcanza 90% en aquellos con canal AV. Se puede decir que los defectos con gran cortocircuito o cardiopatías complejas tienen un mayor riesgo de desarrollar en forma temprana, durante la niñez, síndrome de Eisenmenger, en comparación con los defectos como la comunicación interauricular o la conexión anómala parcial de venas pulmonares cuya presentación es en la edad adulta.
Muchos de los esfuerzos en el campo de la cardiología pediátrica han estado enfocados en evitar la hipertensión pulmonar, pero aun un grupo importante de pacientes que son sometidos a cirugía correctiva en forma tardía la presentan. Los pacientes con HAP pueden tener problemas significativos en el perioperatorio de cirugía no cardiaca, por lo que deben ser evaluados en forma minuciosa para evitar factores que puedan condicionar crisis hipertensivas; de existir diversos factores de riesgo tendrán que ser canalizados a instituciones con experiencia en el manejo de este tipo de pacientes. El síndrome de Eisenmenger es la causa más común de cianosis secundaria a cardiopatía congénita en el paciente en edad adulta.
La hipertensión pulmonar que condiciona cortocircuito inverso de derecha a izquierda se desarrolla a diferente ritmo, dependiendo de la anomalía cardiaca subyacente. En el caso de comunicación interventricular amplia el escenario es que se produzca en la niñez, pero en presencia de comunicación interauricular en su mayoría se desencadena en la edad adulta. Una vez que se establecen los cambios histopatológicos en la vasculatura pulmonar la calidad de vida se va deteriorando y disminuye la esperanza de vida, si bien, en algunos pacientes, se ha reportado sobrevida después de los 60 años. En el tratamiento de la HAP asociada a CC, con los fármacos empleados se ha intentado modificar las tres vías fisiopatológicas conocidas, se agrupan en prostanoides, antagonistas de receptores de endotelina-1 e inhibidores de la fosfodiesterasa.17
Las guías de la Sociedad Europea de Cardiología recomiendan en los pacientes con síndrome de Eisenmenger en clase funcional III la utilización de bosentán, antagonista no selectivo de los receptores de endotelina, sustentado en el primer estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo, el Bosentan Randomised Trial of Endothelin Antagonist Therapy (BREATHE-5). En él se evaluaron durante 16 semanas la saturación de oxígeno, aspectos hemodinámicos y la capacidad de ejercicio, en pacientes con síndrome de Eisenmenger. Los resultados iníciales mostraron una disminución de la presión media pulmonar, mejoría en el índice de resistencia vascular pulmonar e incremento en la capacidad de ejercicio; la prolongación del estudio a 24 semanas mostró que dichos beneficios se mantenían. Posteriormente, diferentes investigaciones han confirmado esta respuesta. En cuanto a los inhibidores de la fosfodiesterasa 5, tanto el sildenafil como el tadalafil han mostrado mejorar la capacidad funcional, el puntaje de Borg y aspectos hemodinámicos en pacientes con HAP secundaria a CC y síndrome de Eisenmenger.17
La realización de cualquier procedimiento quirúrgico en pacientes adultos con cardiopatía congénita conlleva un riesgo. En el transcurrir de la vida de este tipo de pacientes a menudo se requieren diversas cirugías para el manejo de litiasis vesicular, apendicitis, escoliosis o bien problemas neurológicos como absceso cerebral. En estos casos se recomienda que sean evaluados preoperatoriamente mediante electrocardiograma, radiografía de tórax, ecocardiograma, biometría hemática y pruebas de coagulación. Los pacientes de riesgo elevado deben ser canalizados a un servicio especializado. Se consideran pacientes de alto riesgo aquellos con cirugía tipo Fontan, hipertensión pulmonar, cianosis importante, cardiopatías complejas con defectos residuales o que requieren de anticoagulación, o bien pacientes con arritmias potencialmente letales. En este grupo de pacientes el manejo anestésico es cardinal, ya que un inadecuado manejo de las resistencias vasculares puede agravar la hipoxemia y condicionar un estado de choque. El adecuado control de líquidos es crítico en pacientes con cardiopatías cianóticas o con fisiología univentricular, así como en los que cursan con insuficiencia cardiaca.
En la evolución de una cardiopatía congénita operada puede existir la indicación de trasplante cardiaco; lamentablemente los pacientes que han sufrido reoperaciones paliativas o correctivas, no tienen el mismo resultado que la población general en el transplante cardiaco, siendo los de peor resultado los pacientes con fisiología univentricular sometidos a cirugía de Fontan.18
Diversas facetas tienen que ser consideradas en las mujeres con cardiopatía congénita que quieren embarazarse o ya lo estén. Es conveniente informar sobre los riesgos potenciales de que el producto curse con alguna cardiopatía congénita, del riesgo de prematuridad (prematurez), bajo peso o de las posibles complicaciones en la madre. En todas las mujeres portadoras de cardiopatía congénita se deben investigar antecedentes de teratógenos y ofrecerles consejos relacionados con la etiología, la herencia, los riesgos de recurrencia y la opinión diagnóstica prenatal. Los futuros padres deben ser informados del riesgo de recurrencia de la cardiopatía congénita, riesgo que depende del tipo de lesión en alguno de los padres o de los descendientes. La posibilidad de recurrencia de la cardiopatía congénita cuando un descendiente está afectado es de 2.3% y se eleva a 7.3 si existen dos descendientes con dicha patología. Por otra parte, si la madre es portadora de una cardiopatía congénita el riesgo de un producto afectado es de 6.7% y si se trata del padre, de 2.1%.19,20
Si bien el número de casos no es importante, el embarazo en el paciente adulto con cardiopatía congénita puede causar una significativa morbimortalidad no obstétrica en la madre y el feto. El embarazo condiciona cambios hemodinámicos importantes, lo cual bajo un contexto de la cardiopatía congénita puede ocasionar una descompensación cardiaca en la madre y la muerte del feto. Además de los cambios hemodinámicos impuestos por el propio embarazo, durante el parto pueden darse factores, como la vasodilatación periférica inducida por la anestesia o la pérdida de sangre, que agraven la disfunción cardiaca en mujeres con una cardiopatía subyacente importante. Idealmente, antes de ocurrir el embarazo debe ser establecido un plan de control y manejo ante los diversos escenarios posibles. Si bien la gestación y el parto pueden cursar sin contratiempos en pacientes adultos con cardiopatías congénitas con buena clase funcional y función ventricular conservada, no ocurre así en pacientes con hipertensión pulmonar donde la presión pulmonar representa ≥ 70% en relación con la presión sistémica, circunstancia en la cual el riesgo de mortalidad materna es muy elevado.
Para evaluar el riesgo materno de complicaciones cardiovasculares se han elaborado diversos puntajes sustentados en estudios de poblaciones con diferentes tipos de cardiopatías congénitas. Los cambios fisiológicos del embarazo pueden provocar morbilidad y mortalidad materna. En los últimos dos decenios la comprensión del riesgo de embarazo y la manera de cuidar a las mujeres con enfermedad cardiaca durante el embarazo han evolucionado. El estudio multicéntrico CARPREG fue el primero en desarrollar un índice de riesgo para predecir la probabilidad de complicaciones cardiacas maternas. El índice de riesgo CARPREG3 se basó en características clínicas y ecocardiográficas, ha sido de los más utilizados. Está compuesto por cuatro datos clínicos y a cada uno se le asigna un punto. Al final, el riesgo se estratifica en cero, uno y más de un punto que corresponden a 5, 27 y 75%, respectivamente. Después, la Organización Mundial de la Salud propuso un índice de riesgo que incluye las cardiopatías congénitas que contraindican un embarazo, por ejemplo en el grupo IV hay la posibilidad de desenlaces fatales en la madre y el feto hasta en 50%. Finalmente, en el CARPREG II se procedió a combinar lesiones específicas y factores generales que al parecer proporcionan una mejor precisión predictiva.21,22
La necesidad de llevar a cabo anticoagulación, aunque no es contraindicación para el embarazo, condiciona un mayor riesgo en el feto y la madre. Debemos recordar que hay diversos medicamentos cardiovasculares que están contraindicados durante la gestación, como los inhibidores de la enzima convertidora de la angiotensina y bloqueadores de los receptores de angiotensina. En el caso de la warfarina, debe utilizarse luego de que se haya platicado en forma extensa con la paciente acerca de los riesgos potenciales durante la gestación.
Aunque existen pocos estudios sobre la seguridad de la utilización de anticonceptivos en las pacientes con cardiopatías congénitas, basta recordar que representan un mayor riesgo, los estrógenos incrementan el riesgo de tromboembolismo en presencia de cianosis, de cirugía para cardiopatías con fisiología univentricular, de fibrilación auricular o bien de hipertensión pulmonar. En mujeres con cardiopatía congénita cianótica se ha aconsejado utilizar métodos de barrera, como la salpingorrafia y el levonorgestrel, el anticonceptivo del día siguiente.
Si bien los avances en el diagnóstico y tratamiento de las cardiopatías congénitas permiten una mayor y mejor supervivencia, esta población representa un problema creciente que podría convertirse en un problema social. Es necesario promover el desarrollo de centros especializados con recursos humanos idóneos en el manejo de la patología congénita del adulto. Se requiere de nuevos programas de entrenamiento del personal médico en centros de alta referencia ya existentes, en la atención de pacientes adultos con cardiopatía congénita. Es de primordial importancia el que este tipo de pacientes cuenten con atención médica adecuada y empleos acordes a su capacidad intelectual y funcional, para lo cual es necesaria la elaboración de guías de práctica clínica adecuadas a nuestra realidad, así como desarrollar un plan estratégico a favor de esta población.